
Abstract
Choroideremia is a bilateral and progressive X-linked inherited disease characterized by widespread chorioretinal atrophy with relative sparing of the macular region. It is caused by mutations in the ubiquitously expressed CHM gene, which lead to the absence of the Rab escort protein 1 (REP-1), resulting in prenylation deficiency. Typical fundus appearances for choroideremia were found in 3 probands from three unrelated Chinese families in our study. We firstly used the targeted exome sequencing (TES) technology to detect mutations in CHM gene. Based on an established filtering strategy of data analyses, along with confirmation by co-segregation, a previously reported mutation (c.1584_1587del TGTT, p.V529Hfs*7) was identified in one family, while two novel mutations (c.227_232delinsTGTCATTTCA, p.Q76Lfs*7; c.710dupA, p.Y237_S238delinsX) were identified in the other two families. These findings not only expands the currently limited spectrum of Chinese disease-causing variants in CHM gene, but also increases our understanding of the phenotypic and genotypic correlations of choroideremia, and may potentially lead to improved genetic counseling and specific treatment for families with choroideremia as well.
Similar content being viewed by others


Introduction
Choroideremia (CHM; MIM 303100) is an X-linked recessive eye disorder, with the incidence ranging from 1:50,000 to 1:100,0001,2. It commonly manifests as a progressive-inherited chorioretinal degenerative disease characterized by a bilateral, diffuse atrophy of the choriocapillaris, retinal pigment epithelium (RPE), and photoreceptors3. Affected males develop night blindness in late childhood, which progresses with a gradual loss of peripheral vision. Normally, their vision deterioration can reach legal blindness by middle age. Female carriers generally have normal vision, visual fields, color vision, and electroretinograms (ERGs); however, due to random X-inactivation (lyonization), patchy areas of chorioretinal degeneration can occur in women4. In both men and women, central vision is typically maintained until late in the progression of the disease.
In patients with choroideremia, diagnoses are confirmed with the identification of mutations in the CHM gene, which is mapped to chromosome Xq21.23. The CHM gene spans over 180 Kb on the X chromosome, and is expressed in many tissue types including retinal photoreceptor, choroid, RPE, and lymphocyte tissues. The mRNA consists of 15 exons that are conserved in transcription. All of the conserved exons, except exon 15 (3,642 bp), are fewer than 400 bp long5. Furthermore, the absence of Rab escort protein-1 (REP-1) encoded by CHM, which results in prenylation deficiency, has been verified as the cause of retinal degeneration in choroideremia6. As an intracellular protein of 653 amino acids and the A component of Rab geranylgeranyltransferase (GGTase), REP-1 plays an important role in the post-translational Isoprenyl modification of Rab proteins as well as in assisting the intracellular vesicular trafficking process7,8. In all tissues, with the exception of the eye, the loss of functioning REP-1 appears to be compensated by REP-29. Consequently, REP-1 function seems to be a vital factor for the function of the retinal pigment epithelium and photoreceptors.
Almost all reported cases of choroideremia so far have been attributed to functionally null mutations combined with the slow rate of degeneration and small size of the CHM protein coding sequence, which make gene therapy with adeno-associated viral (AAV) vectors an appealing treatment strategy. The subsequent preclinical data for the effects of the vector (AAV2.REP1) in restoring prenylation activity to human choroideremia fibroblasts and initial findings from a phase 1/2 clinical trial have been described10,11.
In published literatures, a wealth of disease-causing mutations have been reported, including nonsense mutations, missense mutations, frameshifts, splice site defects, gross insertions, small deletions, and deletion of the entire CHM gene. More than 100 different pathogenic CHM mutations are currently documented in the RetinoGenetics12. In spite of the large existing spectrum of disease-causing variants in CHM gene, the investigations of Chinese patients with choroideremia are rather limited. We herein describe the phenotype and genotype of members in three Chinese families with choroideremia, and disclose the detection of novel mutations of the CHM gene firstly with the targeted exome sequencing (TES) technology.
Materials and Methods
Subjects and phenotyping
The study protocol was in accordance with the tenets of the Declaration of Helsinki and approved by the Ethics Committee of The Eye Hospital of Wenzhou Medical University. Informed consent was obtained from each participant. Three probands and two of their family members were enrolled in this study. Each of them underwent a comprehensive ophthalmologic examination comprising best-corrected visual acuity (BCVA), slit-lamp biomicroscopy, and color vision screening, which was supplemented with a series of imaging studies including fundus photography and optical coherence tomography (OCT). A panel of ophthalmologists and optometrists reviewed all ocular findings, and confirmed the clinical diagnosis of choroideremia in affected individuals. We contacted all of the genetically related family members, and obtained a detailed family history from each of that were recruited. Moreover, the five participants donated their blood for molecular studies. Physical examinations were performed to exclude systemic diseases, and no other remarkable diseases were found in the subjects’ medical histories.
Library preparation and Targeted exome sequencing
Genomic DNA was extracted from peripheral blood using standard organic extraction procedures. The DNA samples were quantified using a Nanodrop 2000 (Thermal Fisher Scientific, DE), and stored below −20 °C until experimental use. Libraries were prepared in the light of Illumina standard protocol. Briefly, a minimum of 3 μg of the DNA was sheared into 350- to 400-bp fragments. The fragmented DNA was end-repaired, a single adenine base was added to 3′ end, Illumina adapters were then ligated to the fragments, and the final products were PCR amplified. The probes, aiming to be hybridized in solution with the target library for TES experiments, were designed to potentially tile along 164 known causative inherited retinal dystrophy (IRD) genes, including one choroideremia-related gene. Afterwards, Illumina sequencing was performed in according to a previous study13.
Bioinformatics analysis
Sequencing reads were aligned to human hg19 reference using BWA (http://bio-bwa.sourceforge.net/), and afterwards, base quality recalibration and local realignment were conducted using the GATK program (http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page)14,15. To filter out common SNPs and InDels, with minor allele frequency (MAF) > 0.01, the Exome-assistant program (http://122.228.158.106/exomeassistant) was then used for annotation of all identified variants. MagicViewer was applied to view the short read alignment and ascertain the candidate SNPs and InDels. Missense variants were assessed with PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), while the splice-site effect was predicted using the BDGP (http://www.fruitfly.org/seq_tools/splice.html) and ASSP programs (http://wangcomputing.com/assp/index.html).
PCR and direct Sanger sequencing
The DNA samples from the 5 participants in this study, as well as 200 unrelated Chinese control individuals, were analyzed by DNA sequencing. All 15 coding exons of the CHM gene were amplified by PCR. The reagents used for a 50 μl reaction were as follows: 25 μl of 2 × Power Taq PCR MasterMix (BIOTEKE CORPORATION), 2.5 μl each of 10 μM forward and reverse primer, 100 ng of genomic DNA template, and sterile H2O. Amplification was performed in a thermal cycler (Applied Biosystems Veriti 96-Well) with the following PCR protocol: initial DNA denaturation occurred at 95 °C for 5 min, 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 60s, and a final extension phase at 72 °C for 7 min. Afterward, the temperature was held at 4 °C. PCR products were subjected to agarose gel electrophoresis, excised from the gel, and subsequently sequenced. The sequencing results were analyzed using FinchTV (version 1.4.0), and compared with the NCBI database reference sequences. Sanger sequencing and co-segregation analyses in the family member samples confirmed the candidate variants.
Results
Clinical features
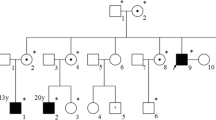
The inheritance patterns of the three Chinese families were all X-linked, as indicated by the familial pedigrees (Fig. 1). The proband F1:IV:2 complained of a long history of night blindness, a progressive decrease in his peripheral vision of each eye, and difficulty with color discrimination. He had normal corneas and anterior chambers, while his lenses were a little turbid (+), which revealed a mild cataract condition. Additionally, he was affected with metamorphopsia. Color vision screening with Ishihara color plates disclosed his limited ability only to identify the test plates in each eye. Fundus exam showed a scalloped border of atrophic changes with easily visible radial choroidal vessels near the macula. There was a residual island of healthy tissue in the central macula and fovea. The optic disc was normal with peripapillary pigment atrophy and the retinal vessels were attenuated (Fig. 2A). OCT images presented a central macular thinning of the retinal outer layers, as well as a discordant reflection pattern within the RPE layer and the choroicapillary layer of each eye, and a discontinuity was found in the retinal neurosensory layer of the left eye (Fig. 2B). The clinical characterizations of the other two probands F2:III:5 and F3:III:5, were very similar to that of the proband F1:IV:2. So was that of the affected male F3:III:4. The female carrier F1:V:2, however, seemed to be asymptomatic. The results for the optometry check of the five participants were shown in Table 1.

Pedigrees of three Chinese families with choroideremia.
Squares and circles indicate males and females, respectively. Rhombus refers to the infant with unknown gender. Deceased family members are represented with a diagonal line, and darkened symbols represent the affected members. The patients below the arrow are the probands.

Imaging studies of the proband F1:IV:2.
(A) Fundus photography showing widespread confluent areas of nummular RPE and choriocapillaris atrophy in the midperiphery and in the paramacular area, sparing only the centermost macular region. (B) OCT images showing reduction of retinal thickness and anomalous reflectivity corresponds well with relative chorioretinal atrophy. RE, right eye; LE, left eye.
Candidate mutations identified by TES
We designed a capture panel to enrich the target DNA by selecting 164 known IRD-related genes from RetNet and OMIM. The enriched DNA was then sent for TES. More than 96.9% of the targeted disease gene regions were sequenced, with an average sequencing depth of 104.38. A total number of 650 variants were identified in the samples. Among them, there were 386 non-synonymous variants, including missense, nonsense, and splicing variants. This number was narrowed down to 46 variants by excluding variants reported in dbSNP137, Exome Variant Server, and ExAC that had minor allele frequencies of (MAF) > 0.01. For missense variants, computational prediction by PolyPhen and SIFT as well as imposing restriction such as the consistency of genetic transmission mode were applied to further reduce the number of candidate mutations. The remaining variants were likely to cause the disease in patients and were further subjected to validation by Sanger sequencing.
Confirmation and co-segregation testing
After a comprehensive screening for mutations by Sanger sequencing and co-segregation analyses of the three Chinese pedigrees, three frameshift mutations in CHM gene were detected, none of which occurred in the 200 controls (Fig. 3A and Supp. Fig. 1). In family 1, the proband F1:IV:2 harbored a hemizygous mutation (c.227_232delinsTGTCATTTCA. p.Q76Lfs*7) within exon 4, which had not been reported and thus was ascertained to be novel. However, his pregnant daughter F1:V:2 was heterozygous for the mutation, while the amniotic fluid was finally confirmed to be wildtype, showing she was carrying a healthy infant. In family 2, a previously reported mutation (c.1584_1587del TGTT. p.V529Hfs*7) within exon 13 was detected16,17,18. In family 3, the proband F3:III:5 and his brother F3:III:4 both harbored a novel hemizygous mutation (c.710dupA, p.Y237_S238delinsX) within exon 6. The three mutations were predicted to stop the open reading frame at codon 82, codon 535, and codon 237, respectively, thereby generating three different prematurely truncated CHM proteins (Fig. 3B).

Mutational analyses of the three Chinese families.
(A) Sequencing results of the family members recruited. Compared to the controls, the male patient F1:IV:2 and the female carrier F1:V:2 from family 1 have an identical 6-bp deletion with a 10-bp insertion mutation, the male patient F2:V:5 from family 2 has a 4-bp deletion mutation, and the two male patients F3:III:5 and F3:III:4 from family 3 have the same duplication mutation. (B) Multiple sequence alignment of CHM polypeptides of different species showing the conserved amino acid residues. The amino acid residues highlighted in bold in the red boxes are the residues after frameshift mutation.
Discussion
Choroideremia occurs in about 4% of the blind population, and has become the second leading cause of hereditary and progressive bilateral night blindness after retinitis pigmentosa. In this study, we reported three Chinese families with choroideremia and shed light on the phenotypic and genotypic defects present in the family members.
The probands showed severe degeneration in their visual acuity when observed in their ophthalmic examinations, and widespread chorioretinal atrophies were seen using fundoscopy and optical coherence tomography scans. A notable difference, in comparison with other retinal degenerative diseases, is that all target tissue layers, including the choroid, RPE, and photoreceptors, are continually shrinking in choroideremia11. Due to the deficiency in the choroid and the fragility of the blood-retina barrier, the affected eyes tend to be vulnerable to other various diseases, which results in direct or indirect damages19. Patients with choroideremia have been reported to have complications involving cataracts20, recurrent uveitis21, and cystic macular edema22. Clinical findings from the affected male F1:IV:2 in our study included poor visual acuity, cataracts, metamorphopsia, and decreased color vision, in addition to the chorioretinal atrophy consistent with the choroideremia diagnosis. Despite these clinical findings, the female carrier F1:V:2 was asymptomatic.
Mutations in the CHM gene cause choroideremia in multiple ethnic groups. Previous studies found that cases of choroideremia had been mostly reported in European and Japanese families, but reported CHM mutations from Chinese families have been limited (Fig. 4). Yip et al.23 reported five mutations in five separate Chinese families, Zhou et al.24 detected three mutations in three mainland Chinese families, Lin et al.19 identified two mutations in two Chinese families, and Guo et al.25 disclosed one frameshift mutation in a Chinese family. From our experimental analyses of three unrelated Chinese families, we expand the spectrum of choroideremia-related mutations by adding two novel mutations (c.227_232delinsTGTCATTTCA, p.Q76Lfs*7; c.710dupA, p.Y237_S238delinsX) and a previously reported mutation (c.1584_1587del TGTT, p.V529Hfs*7) to the existing disease-causing variants in CHM gene.

Schematic diagram of the mutations in the CHM gene.
The black and white bars of various lengths with numbers below them represent different exons. The longitudinal bars indicate different types of mutations within different exons and flanking introns, with the red bars showing substitution mutations, the green bars showing frameshift mutations, and the purple bars showing splicing mutations. The number of the colored longitudinal bars corresponds with the number of reported mutations found in multiple ethnic groups. The mutations occur in different ethnicities are noted by the vertical line, black patch, horizontal lines, left diagonals, right diagonals and black dots inside the bars, respectively representing American, Chinese, European, Japanese, Oceanian, and other nationalities. The gray transverse bars in the top tier indicate the GDI domains.
In this work, candidate variants were only considered pathogenic when they satisfied following criteria: (1) missense mutations were positioned in the amino-acid conserved region across species, (2) splice-site variations fulfilled the GT-AT rules, (3) stop/frameshift variants were present, and (4) the mutations were predicted to be damaging or disease-causing by no less than two of the bioinformatic programs (PolyPhen-2, SIFT, MutationTaster, and ASSP)26. The novel mutations found in family 1 and family 3 of our study both met the third criterion, and were further verified to cause the premature translation of the protein. As a result, they were critically confirmed to be pathogenic equivalents to the known mutation detected in family 2 (Table 2). Most of the CHM mutations result in absence of REP-1 due to premature termination codons (PTCs) and degradation of the inappropriately folded protein or truncated mRNA27,28. Our data demonstrated three different types of truncations in the CHM gene, producing a truncated REP-1 protein of 81 aa, 534 aa, and 236 aa, respectively. The truncated REP-1 protein was assumed to be degraded enzymatically in vivo in the affected members of these CHM families.
In summary, our study uncovered the clinical and genetic characteristics of three Chinese families with choroideremia. The primary methods used to identify disease-causative genes are currently positional cloning and linkage analysis. Nevertheless, there are tremendous limitations within the conventional strategies. Recently, targeted exome sequencing (TES) technology has become an efficient for exploring disease-related genes29. We firstly used the TES approach to successfully identify mutations in CHM from pedigrees with choroideremia, including two novel mutations as well as a known causative mutation. Considering the developing retinal gene therapy for choroideremia, this work will not only increase our understanding of the genetic etiology of choroideremia, but may potentially lead to improved genetic counseling and specific treatment for families with choroideremia as well.
Additional Information
How to cite this article: Cai, X.-B. et al. Novel CHM mutations identified in Chinese families with Choroideremia. Sci. Rep. 6, 35360; doi: 10.1038/srep35360 (2016).
References
Coussa, R. G. & Traboulsi, E. I. Choroideremia: a review of general findings and pathogenesis. Ophthalmic Genet 33, 57–65 (2012).
MacDonald, I. M., Sereda, C., McTaggart, K. & Mah, D. Choroideremia gene testing. Expert Rev Mol Diagn 4, 478–484 (2004).
Cremers, F. P., van de Pol, D. J., van Kerkhoff, L. P., Wieringa, B. & Ropers, H. H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 347, 674–677 (1990).
Rudolph, G. et al. Phenotypic variability in three carriers from a family with choroideremia and a frameshift mutation 1388delCCinsG in the REP-1 gene. Ophthalmic Genet 24, 203–214 (2003).
van Bokhoven, H. et al. Cloning and characterization of the human choroideremia gene. Hum Mol Genet 3, 1041–1046 (1994).
Seabra, M. C., Brown, M. S. & Goldstein, J. L. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science 259, 377–381 (1993).
Pereira-Leal, J. B., Hume, A. N. & Seabra, M. C. Prenylation of Rab GTPases: molecular mechanisms and involvement in genetic disease. FEBS Lett 498, 197–200 (2001).
Goody, R. S., Rak, A. & Alexandrov, K. The structural and mechanistic basis for recycling of Rab proteins between membrane compartments. Cell Mol Life Sci 62, 1657–1670 (2005).
Seabra, M. C., Ho, Y. K. & Anant, J. S. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 270, 24420–24427 (1995).
Tolmachova, T. et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med (Berl) 91, 825–837 (2013).
MacLaren, R. E. et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 383, 1129–1137 (2014).
Ran, X. et al. ‘RetinoGenetics’: a comprehensive mutation database for genes related to inherited retinal degeneration. Database (Oxford) 2014 (2014).
Huang, X. F. et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med 17, 271–278 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
van den Hurk, J. A. et al. Detection and characterization of point mutations in the choroideremia candidate gene by PCR-SSCP analysis and direct DNA sequencing. Am J Hum Genet 50, 1195–1202 (1992).
Schwartz, M., Rosenberg, T., van den Hurk, J. A., van de Pol, D. J. & Cremers, F. P. Identification of mutations in Danish choroideremia families. Hum Mutat 2, 43–47 (1993).
Li, S. et al. Exome sequencing reveals CHM mutations in six families with atypical choroideremia initially diagnosed as retinitis pigmentosa. Int J Mol Med 34, 573–577 (2014).
Lin, Y. et al. Molecular analysis of the choroideremia gene related clinical findings in two families with choroideremia. Mol Vis 17, 2564–2569 (2011).
Binkhorst, P. G. & Valk, L. E. A case of familial dwarfism, with choroideremia, myopia, posterior polar cataract, and zonular cataract. Ophthalmologica 132, 299 (1956).
O, S. J., Kim, S. H. & Lee, H. Y. A case of choroideremia with recurrent anterior uveitis. Korean J Ophthalmol 17, 55–62 (2003).
Genead, M. A. & Fishman, G. A. Cystic macular oedema on spectral-domain optical coherence tomography in choroideremia patients without cystic changes on fundus examination. Eye (Lond) 25, 84–90 (2011).
Yip, S. P. et al. Novel truncating mutations of the CHM gene in Chinese patients with choroideremia. Mol Vis 13, 2183–2193 (2007).
Zhou, Q. et al. Genetic and phenotypic characteristics of three Mainland Chinese families with choroideremia. Mol Vis 18, 309–316 (2012).
Guo, H. et al. Whole-exome sequencing reveals a novel CHM gene mutation in a family with choroideremia initially diagnosed as retinitis pigmentosa. BMC Ophthalmol 15, 85 (2015).
Furgoch, M. J., Mewes-Ares, J., Radziwon, A. & Macdonald, I. M. Molecular genetic diagnostic techniques in choroideremia. Mol Vis 20, 535–544 (2014).
Sergeev, Y. V. et al. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res 665, 44–50 (2009).
Iino, Y., Fujimaki, T., Fujiki, K. & Murakami, A. A novel mutation (967 − 970 + 2)delAAAGGT in the choroideremia gene found in a Japanese family and related clinical findings. Jpn J Ophthalmol 52, 289–297 (2008).
Sun, Y. et al. A novel mutation of EYA4 in a large Chinese family with autosomal dominant middle-frequency sensorineural hearing loss by targeted exome sequencing. J Hum Genet 60, 299–304 (2015).
Acknowledgements
We thank the patients and their family members for their participation in this study. This study was supported by the National Key Basic Research Program (2013CB967502 to ZJ), National Natural Science Foundation of China (81371059, 81522014 to ZJ), Zhejiang Provincial Natural Science Foundation of China (LR13H120001, LY12H12003), Zhejiang Provincial Key Research and Development Program (2015C03029), NHFPC Grant-in-Aid for Medical and Health Science (201472911), Wenzhou Science and Technology Innovation Team Project (C20150004), National Key Clinical Specialty (Ophthalmology), MOST Projects (2012YQ12008004), the project of Zhejiang Provincial Top Key Discipline and Key Construction Discipline of Medicine, Wenzhou Science and Technology Foundation (Y20140358).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Z.-B.J. and Q.-K.L. Performed the experiments: X.-B.C. and X.-F.H. Analyzed the data: X.-B.C. and X.-F.H. Contributed reagents/materials/analysis tools: Y.T. and Q.-K.L. Wrote the paper: X.-B.C. and Z.-B.J.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cai, XB., Huang, XF., Tong, Y. et al. Novel CHM mutations identified in Chinese families with Choroideremia. Sci Rep 6, 35360 (2016). https://doi.org/10.1038/srep35360
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35360
- Springer Nature Limited
This article is cited by
-
Clinical and genetic findings in a Chinese cohort with choroideremia
Eye (2023)
-
Whole-exome sequencing identified a novel mutation in CHM of a Chinese family
Journal of Genetics (2021)
-
Expanding the genetic spectrum of choroideremia in an Australian cohort: report of five novel CHM variants
Human Genome Variation (2020)
-
Novel CHM mutations in Polish patients with choroideremia – an orphan disease with close perspective of treatment
Orphanet Journal of Rare Diseases (2018)