
Abstract
Congenital long QT syndrome (LQTS) diagnosis is complicated by limited genetic testing at scale, low prevalence, and normal QT corrected interval in patients with high-risk genotypes. We developed a deep learning approach combining electrocardiogram (ECG) waveform and electronic health record data to assess whether patients had pathogenic variants causing LQTS. We defined patients with high-risk genotypes as having ≥1 pathogenic variant in one of the LQTS-susceptibility genes. We trained the model using data from United Kingdom Biobank (UKBB) and then fine-tuned in a racially/ethnically diverse cohort using Mount Sinai BioMe Biobank. Following group-stratified 5-fold splitting, the fine-tuned model achieved area under the precision-recall curve of 0.29 (95% confidence interval [CI] 0.28–0.29) and area under the receiver operating curve of 0.83 (0.82–0.83) on independent testing data from BioMe. Multimodal fusion learning has promise to identify individuals with pathogenic genetic mutations to enable patient prioritization for further work up.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Congenital long QT syndrome (LQTS) is a genetic condition that impacts 1 out of 2000 lives. It leads to dysfunctional cardiac repolarization and manifests as sudden cardiac arrest in 13% of patients1,2. LQTS is defined by a prolongation of the cardiac action potential, reflected as a corrected QT (QTc) interval >440 ms among males or >460 ms among females3. This pattern can lead to irregular heartbeat pattern, torsades de pointes, and potentially life-threatening ventricular arrhythmias, sometimes as the first symptom of the disease.
Congenital LQTS has several genetic variants implicated. Specifically, mutations in three major genes have been causally linked: KCNQ1 in subtype 1, KCNH2 in subtype 2, and SCN5A in subtype 32. However, widespread genetic testing of the general population is currently limited. Routine clinical genomic testing, while desirable, is complicated by cost, logistics and need for expert interpretation at a large scale, especially when individual experiences and perceptions of arrhythmias vary widely4.
According to the Schwartz score, diagnosis and risk stratification depend on clinical and family history and electrocardiogram (ECG) features1,5. There are a few challenges with the utility of this criteria. One major issue in the diagnosis of LQTS is symptom variability, which can sometimes result in asymptomatic presentation6. The arrhythmias associated with LQTS may be triggered by various factors such as stress, physical activity, or pharmacotherapy, adding complexity to the diagnosis as symptoms may not be consistently present and perceived6.
In addition, cases of LQTS may lack observable ECG characteristics. QT interval and T wave morphology constitute key criteria for the Schwartz score, yet normal QTc in up to 40% of patients with genetic risk can complicate detection of the disease3. While these patients have a lower risk of life-threatening events compared to those with evident symptoms, their risk is still higher than unaffected genotype-negative family members1,7. These patients also remain predisposed to developing life-threatening torsades de pointes if exposed to pharmacological substances that inhibit potassium channels and may pass on phenotypic disease to future generations8. Above all, even if patients demonstrate long QTc, this presentation often fails to trigger work up because of the multiple etiologies of QTc prolongation. To better improve the use of information from waveform data, artificial intelligence could be leveraged on ECGs for genomic inference.
Finally, LQTS has been largely understudied in minority populations as most publications have been conducted in White populations. Black individuals have more severe disease with longer QTc intervals and more frequent cardiac events than White patients9. There also exist significant differences in the prevalence of channel variants in potassium channel genes across Asian, Black, Hispanic, and White ethnic groups10. Various genetic forms of LQTS have been linked to unique ECG characteristics that correspond to type and extent of ion channel abnormalities11. Thus, there is a critical need for scalable approaches to identify patients with pathogenic variants using easily available clinical data modalities such as the electrocardiogram (ECG).
Previously, models have used derived ECG features such as RR-interval, QTc-intervals, and T-wave ECG intervals to identify genotype-positive individuals12,13. Models have additionally been developed using deep learning (DL) approaches to leverage ECG morphology to infer genotype status for LQTS14,15,16. However, among studies with published information on patient background, models were only developed on patients of European ancestry. Moreover, none have a combination of ECG waveform and corresponding electronic health record (EHR) data.
To facilitate LQTS diagnosis, we use multimodal fusion modeling using ECG and EHR phenotypes to indicate genotype positivity for subtypes 1 and 2 in a DL model. This algorithm fuses waveform and clinical data to detect genotype-positive patients for pathogenic variants causing LQTS.
Results
We pre-trained the fusion model using the United Kingdom Biobank (UKBB)17 (Table 1). The median age was 54 years, 49% were female, and 96% were White. LQTS subtypes 1 and 2 were represented: 45 patients had variants associated with subtype 1, and 7 patients had variants associated with subtype 2. We included 59 ECGs from genotype-positive patients and 1181 ECGs from genotype-negative patients. We then fine-tuned the model on the Mount Sinai BioMe Biobank. Median age was 64 years, 12 were female, and 1 was American Indian, 4 Black, 4 Hispanic. No patients were of European ancestry. LQTS subtypes 1 and 2 were represented: 4 patients had variants associated with subtype 1, 11 patients had variants associated with subtype 2, and 2 patients had variants associated with both. We included 49 ECGs from genotype-positive patients and 730 ECGs from genotype-negative matched controls.
The model pretrained on UKBB data had an area under the precision-recall curve (AUPRC) of 0.23 (95% confidence interval [CI] 0.22–0.24, improvement in the baseline prevalence of 0.05) and an area under the receiver operating curve (AUROC) of 0.76 (0.75–0.77) on hold-out UKBB data (Fig. 1a). After further fine-tuning on BioMe, the fine-tuned model had AUPRC of 0.29 (0.28–0.29) and AUROC of 0.83 (0.82–0.83) on hold-out data (Fig. 1b).

a Model performance of the baseline model using data from the UKBB. b Model performance of the fine-tuned model using data from Mount Sinai BioMe Biobank. Light blue shading around the plotted lines indicates confidence interval.
We performed chart review among genotype-positive patients in BioMe to ascertain the utility of this approach in identifying patients with pathogenic variants (Table 2). We found no patient had received subsequent clinical evaluation or workup. Four of 16 genotype-positive patients were concurrently prescribed a QT prolonging medication at time of ECG acquisition, with two of these patients demonstrating a prolonged QTc. Ten of 16 genotype-positive patients had a normal QTc on ECG, indicating the difficulty of diagnosing this disease based on ECG alone. Of the genotype-positive patients, 2/16 (~13%) had an emergency presentation for syncope, presyncope, or ventricular arrhythmia.
We then used this model to identify patients in the greater Mount Sinai Health System who did not have diagnoses for LQTS but could potentially be genotype-positive (Fig. 2). We randomly selected 11,446 patients who were not included in previous model development and testing and did not possess prior genetic testing. We investigated the probabilities returned by the model. The mean probability is 0.060, with standard deviation of 0.10, and the median probability is 0.016. The range is 0 to 0.706, and the 25th, 50th, and 75th percentiles were 0.0035, 0.016, 0.062, respectively. We examined clinical history and ECG waveform of the patients with the highest probabilities of LQTS. Their probabilities ranged from 0.642 to 0.706.

Patients without diagnosis of congenital long QT syndrome (LQTS) were randomly selected from the Mount Sinai Healthcare system. A board certified electrophysiologist evaluated patients identified by the model has having either high or low probability of pathogenic variants for LQTS.
Among the ten patients with the highest probabilities of LQTS identified by the model, six had prolonged QTc. Two of these six patients had history and ECG presentations that were suggestive of congenital LQTS, as identified by a board-certified cardiac electrophysiologist (Table 3). A representative ECG is included (Fig. 3). We further examined the ECGs of the next ten patients with the highest probability of LQTS or those in the top 11–20, as well as those with the lowest probability of LQTS as identified by the model. We found no evidence of LQTS according to clinical or ECG presentation, suggesting that the sensitivity of our model is limited to the top ten patients by probability ranking.

The morphology of QTc prolongation is consistent particularly with LQTS2 with broad, notched T waves.
Finally, we implemented Captum, a Python library for machine learning model explainability, to determine contributions of features to model predictions. Output decisions were attributed 69.44% to clinical variables and 30.56% to ECG. Among clinical variables, age contributed 11.49%, gender 7.47%, ventricular rate 9.20%, atrial rate 1.27%, PR interval 1.25%, and QTc interval 69.33% to model predictions (Fig. 4).

Captum was used to quantify influence on model decision-making by (a) modality and (b) clinical variables.
Discussion
The fusion model demonstrated modest performance when using data from UKBB. However, when fine-tuned to include diverse data, the DL model achieved better performance in predicting pathogenic LQTS variants. As genetic testing is infrequently performed on a population level, especially in minority populations, leveraging fusion models may identify at-risk patients who may benefit from further workup18,19,20. By incorporating a broader range of patient data, the model may better capture variations that exist in different populations and provide more accurate predictions for a broader range of individuals.
Moreover, when the model was assessed on patients in the greater Mount Sinai Health System, two patients among the top 10 highest probabilities were found to possibly have high risk genotypes for congenital LQTS, as suggested by their clinical history or ECG presentation. Given the prevalence of LQTS at an estimated 1:2000, the identification of patients possibly at high risk for LQTS for further work up could be facilitated by this algorithm. This risk stratification would be cost efficient and scalable in comparison to universal genetic testing.
We note that some patients who were genotype positive and included in model training and those identified as having high risk by the model were concurrently prescribed or self-reported using QT prolonging medications. While these pharmacological agents are known to impact QT prolongation, the magnitude of QT prolonging effect may reflect superimposed drug effect on underlying channelopathy and modify arrhythmia risk. Thus, these medications may cause a more profound impact in patients with congenital LQTS that could be dismissed as acquired LQTS. We believe that once identified by this model, these patients could still benefit from further work up to ascertain pathogenic genotype and better characterize arrhythmogenic risk whether in the presence of LQTS alone or confounded by medical therapy.
In the past, models for classifying individuals with LQTS utilized features extracted from ECG. Some models also employed DL approaches to utilize ECG morphology for predicting genotype status12,13. However, among studies with published patient demographics, these models were predominantly developed using data from patients of European ancestry, with a lack of integration between waveform data and clinical data in these studies14,15,16. Multimodal fusion learning allows the incorporation of additional information from EHR with ECG data. By jointly learning from both ECG waveforms and EHR data, the model was able to consider relationships between modalities that are not possible when analyzing each source of data in isolation. This integration provides a more comprehensive view of patient information, improving performance of the model.
The purpose of this model is to aid in early and accurate identification of individuals at risk for LQTS-associated complications. Genotype prediction from commonly used healthcare modalities can inform tailored treatment strategies and risk management plans. Moreover, addressing genetic diversity is crucial for ensuring that diagnostic and predictive tools are effective across different populations, thereby promoting equitable healthcare practices. By accounting for genetic diversity through the inclusion of diverse patient populations, the model is more inclusive and applicable across various ethnic backgrounds.
Our study is limited by small sample sizes and low prevalence of genotype positivity and control under sampling for model training. However, congenital LQTS itself is a rare disease. In addition, we are limited from assigning a definitive, quantitative threshold as this will be influenced by patient population size. Most patients included for model training were identified as having European ancestry. Nonetheless, this model was fine-tuned on one of the most racially and ethnically diverse populations according to previously published work. Moreover, extensive patient and family histories are not consistently available in real world data for the calculation of Schwarz score. Lastly, since the BioMe and UKBB populations are composed of people over 18 years of age, the algorithm’s performance in a younger and potentially higher risk population of patients with LQTS is unknown but will be the subject of future work.
In summary, multimodal AI applied to the ECG and EHR holds potential to identify patients that have known pathogenic mutations implicated in LQTS from diverse racial/ethnic backgrounds. This algorithm can help to prioritize patients for expert referral and guide confirmatory genetic testing pending external validation.
Methods
Genotype identification
The UKBB is a comprehensive biomedical database of participants 40–69 years of age from the United Kingdom, including demographic, ECG, and genotyping data. The Mount Sinai BioMe Biobank comprises over 50,000 participants from diverse racial/ ethnic backgrounds who have sought care in the Mount Sinai Health System in New York City. Genotypic, sequencing, and EHR information are available. In UKBB, 55 patients were genotype positive. For both BioMe and UKBB, all participants were at least 18 years of age. All participates provided informed consent to make their data available for research. This study was approved by the institutional review board at the Icahn School of Medicine at Mount Sinai, and UKBB data use was approved under application #16218 in the United Kingdom Biobank (UKBB).
We identified genotype-positive patients as having ≥1 of 3 known pathogenic variants in a LQTS-susceptibility genes21. In observation of American College of Medical Genetics and Genomics guidelines, all included variants were classified as “pathogenic” or “likely pathogenic.” Variants were confirmed to have minor allele frequency <0.1%. We excluded mutations that were synonymous, intronic, or appearing in untranslated regions, unless previously reported as pathogenic22. Variants were determined to be pathogenic by the LQTS Clinical Domain Channelopathy Working Group; they were subsequently curated through ClinVar23. Results of mutations in individual patients are included in Supplementary Tables 1 and 2.
Data processing
The data are 2D ECG plots and clinical data that are readily accessible from electronic health records, specifically age and sex, and automatic output from ECGs, namely PR interval, QTc interval, atrial rate, and ventricular rate. ECG information was extracted from XML files, which contained raw waveforms in integer representation, extracted ECG features, and demographics. Waveforms were captured at 500 Hz, and the first 5 s of recording was used. Leads I, II, and V1-6 were plotted as 2D image plots. Lead III and augmented unipolar leads aVL, aVR, and aVL were excluded as they can be derived from the limb and precordial leads. A Butterworth Bandpass filter, with cutoff frequencies of 0.5 and 40 Hz, was applied to ECGs to remove recording artifacts because of electrical activity from other sources24. We matched genotype-positive patients with genotype-negative patients for age, sex, and self-reported race/ethnicity in an approximately 1:20 ratio.
Multimodal fusion learning
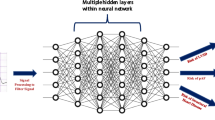
We fused a convolutional neural network (CNN) for ECGs and a Multi-Layer Perceptron (MLP) (Fig. 5)25. This architecture was previously developed for a different task, but model parameters have been completely reinitialized for this study25. The specific convolutional neural network architecture used was the Efficientnet-B4, which was chosen because it has had previous success in image classification models by appropriately scaling input images and neural network dimensions26. Meanwhile, three fully connected layers with non-linear Rectified Linear Activation Units constituted the MLP. The fully connected output layer of the CNN was substituted with another fully connected layer comprising 64 neurons, which was connected to the MLP. The final model hyperparameters included cross entropy loss, Adam optimizer, and a learning rate of 1e−3. Model performance was evaluated based on AUROC and AUPRC.

FC Layer stands for fully connected layer. The number of neurons for each layer of the multi-layer perceptron processing the tabular data are 128, 64, and 32 neurons, respectively. The number of neurons for each layer of the multi-layer perceptron processing the tabular and waveform data are 64, 32, and 16 neurons, respectively, with a classification head composed of 2 neurons.
Transfer learning
Transfer learning can be employed as a technique to leverage knowledge gained from training a neural network on one task and apply it to a related task. This involves first pre-training the model on data that resemble the downstream to adjust weights of the neural network to capture relevant features. Subsequently, the network is fine-tuned using data specific to the downstream task. This is useful to mitigate bias from data derived from a single institution and natural data scarcity in rare diseases, such as in classifying patients with LQTS.
The UKBB and BioMe datasets were selected for their size and relative dissimilarity in the racial/ ethnic backgrounds of patients. The UKBB is composed mostly of patients of European ancestry. Meanwhile, the majority of BioMe are of non-European background, representing patients of greater racial/ ethnic diversity. The weights of all neural network layers were adjusted during fine-tuning, and the same learning rate was used between pre-training and fine-tuning. For both populations, informed consent was obtained from each patient, and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki in a priori approval by the organization’s human research committee.
Cross-fold group stratified training
We a priori set aside 25% of data from the BioMe and UKBB databases for testing. Group stratified 5-fold splitting was selected to ensure that all data points for the same patient are kept together within a fold, maintain comparable class representation across folds, and safeguard model performance for resampling in a small, class-imbalanced dataset. Within each cross fold, data were divided into 80% training and 20% validation data. An early stopping term was set in which training continued until no further improvement was demonstrated on validation data for 3 epochs. The best model was selected based on its performance on testing data following 500 times bootstrapping, or resampling the dataset 500 times with replacement and assessing model performance.
Chart review
Chart review was conducted by two clinicians among BioMe patients with pathogenic variants. All documentation was thoroughly analyzed to determine if patients ever had a previous diagnosis of long QT syndrome. QT intervals of ECG waveform were manually evaluated, and ST segments and T wave morphologies were assessed for known patterns associated particularly with LQTS. Charts were also analyzed for any emergency presentations with presyncope, syncope, or ventricular arrhythmias. Finally, the potential use of QT interval prolonging medications during the time of ECGs were noted27. These medications include antiarrhythmics (amiodarone, dofetilide, flecainide, procainamide, quinidine, sotalol); antibiotics (fluoroquinolones and macrolides); antidepressants (amitriptyline, citalopram, imipramine), antipsychotics (droperidol, haloperidol, olanzapine, quetiapine, risperidone, thioridazine, ziprasidone), and other drugs (cisapride, methadone, ondansetron, sumatriptan)27. Drugs that were prescribed as well as self-reported were included.
Retrospective clinical validation
Files of 11,446 patients who have had ECGs performed, no genotyping, and no formal diagnosis of LQTS were selected from the Mount Sinai Health System. ECG and EHR data were processed as aforementioned and served as input for the fine-tuned model. For each patient, the predicted probability of LQTS was produced. Twenty patients with the highest probabilities and the ten patients with the lowest probabilities were selected for further evaluation of their ECG waveform, prescription or reported use of QT prolonging medications during time of ECGs, and clinical history for any emergency presentations with presyncope, syncope, or ventricular arrhythmias by a board-certified electrophysiologist.
Software
All coding was performed in Python. pandas28, PIL29, Pytorch30, matplotlib31, numpy32, os33, scikit-learn34, seaborn35, scipy36, torchvision37, and captum38 libraries were used. This study was approved by the Mount Sinai IRB. The underlying code can be accessed via this link: https://github.com/jiangj07/lqts.
Data availability
United Kingdom Biobank dataset analyzed during the current study are publicly available, https://www.ukbiobank.ac.uk/enable-your-research/about-our-data. The Mount Sinai BioMe Biobank analyzed during the current study are not publicly available due to restrictions placed by the Mount Sinai Institutional Review Board, but deidentified data are available from the corresponding author on reasonable request.
Code availability
The underlying code for this study is available in Github and can be accessed via this link: https://github.com/jiangj07/lqts.
References
Schwartz, P. J. & Ackerman, M. J. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur. Heart J. 34, 3109–3116 (2013).
Nakano, Y. & Shimizu, W. Genetics of long-QT syndrome. J. Hum. Genet. 61, 51–55 (2016).
Krahn, A. D. et al. Congenital long QT syndrome. Clin. Electrophysiol. 8, 687–706 (2022).
Katsanis, S. H. & Katsanis, N. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 14, 415–426 (2013).
Veltmann, C. & Borggrefe, M. A’Schwartz score’for short QT syndrome. Nat. Rev. Cardiol. 8, 251–252 (2011).
Shah, S. R., Park, K. & Alweis, R. Long QT syndrome: a comprehensive review of the literature and current evidence. Curr. Probl. Cardiol. 44, 92–106 (2019).
Goldenberg, I. et al. Risk for Life-Threatening Cardiac Events in Patients With Genotype-Confirmed Long-QT Syndrome and Normal-Range Corrected QT Intervals. J. Am. Coll. Cardiol. 57, 51–59 (2011).
Priori, S. G., Napolitano, C. & Schwartz, P. J. Low Penetrance in the Long-QT Syndrome. Circulation 99, 529–533 (1999).
Fugate II, T. et al. Long QT Syndrome in African-Americans. Ann. Noninvasive Electrocardiol. 15, 73–76 (2010).
Ackerman, M. J. et al. Ethnic Differences in Cardiac Potassium Channel Variants: Implications for Genetic Susceptibility to Sudden Cardiac Death and Genetic Testing for Congenital Long QT Syndrome. Mayo Clin. Proc. 78, 1479–1487 (2003).
Zareba, W. Genotype-specific ECG patterns in long QT syndrome. J. Electrocardiol. 39, S101–S106 (2006).
Hermans, B. J. et al. Improving long QT syndrome diagnosis by a polynomial-based T-wave morphology characterization. Heart Rhythm 17, 752–758 (2020).
Hermans, B. J. et al. Support vector machine-based assessment of the T-wave morphology improves long QT syndrome diagnosis. EP Europace 20, iii113–iii119 (2018).
Aufiero, S. et al. A deep learning approach identifies new ECG features in congenital long QT syndrome. BMC Med. 20, 1–12 (2022).
Doldi, F. et al. Detection of patients with congenital and often concealed long-QT syndrome by novel deep learning models. J. Personalized Med. 12, 1135 (2022).
Prifti, E. et al. Deep learning analysis of electrocardiogram for risk prediction of drug-induced arrhythmias and diagnosis of long QT syndrome. Eur. Heart J. 42, 3948–3961 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Hann, K. E. J. et al. Awareness, knowledge, perceptions, and attitudes towards genetic testing for cancer risk among ethnic minority groups: a systematic review. BMC Public Health 17, 503 (2017).
Suther, S. & Kiros, G.-E. Barriers to the use of genetic testing: a study of racial and ethnic disparities. Genet. Med. 11, 655–662 (2009).
Grosse, S. et al. Population screening for genetic disorders in the 21st century: evidence, economics, and ethics. Public Health Genomics 13, 106–115 (2009).
Adler, A. et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141, 418–428 (2020).
Lahrouchi, N. et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 142, 324–338 (2020).
Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844 (2020).
Jagtap, S. K. & Uplane, M. in 2012 International Conference on Communication, Information & Computing Technology (ICCICT). 1–6 (IEEE, 2012).
Vaid, A. et al. Automated determination of left ventricular function using electrocardiogram data in patients on maintenance hemodialysis. Clin. J. Am. Soc. Nephrol. 17, 1017–1025 (2022).
Koonce, B. EfficientNet. In Convolutional Neural Networks with Swift for Tensorflow: Image Recognition and Dataset Categorization, 109–123. Apress Berkeley, CA (2021). https://doi.org/10.1007/978-1-4842-6168-2.
Farzam, K. & Tivakaran, V. S. QT prolonging drugs. StatPearls Publishing, Treasure Island (2018).
McKinney, W. & Team, P. Pandas-Powerful python data analysis toolkit, 1625 (2015).
Umesh, P. Image processing in python, 23 (CSI Communications, 2012).
Imambi, S., Prakash, K. B. & Kanagachidambaresan, G. PyTorch. In Programming with TensorFlow: Solution for Edge Computing Applications, 87–104. (Springer Nature Switzerland AG 2021).
Yim, A., Chung, C. & Yu, A. Matplotlib for Python Developers: Effective techniques for data visualization with Python (Packt Publishing Ltd, 2018).
Harris et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Zhang, Y. & Zhang, Y. An Introduction to Python and computer programming (Springer, 2015).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Sial, A. H., Rashdi, S. Y. S. & Khan, A. H. Comparative analysis of data visualization libraries Matplotlib and Seaborn in Python. Int. J Adv. Trends Comp. Sci. Engin. 10, 277–281 (2021).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Marcel, S. & Rodriguez, Y. Torchvision the machine-vision package of torch. Proceedings of the 18th ACM international conference on Multimedia 2010. 1485–1488 (2010).
Kokhlikyan, N. et al. Captum: A unified and generic model interpretability library for pytorch. Preprint at https://arxiv.org/abs/2009.07896 (2020).
Acknowledgements
This project was funded by 24PRE1184156, 5R01HL155915-03, 1TL1TR004420-01, T32GM146636, and UL1TR004419. The funding sources played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
J.J., J.L., and G.N.N. conceptualized the study. J.J. and J.L. completed the literature review. J.J. and A.V. designed the experiments. H.V. identified genotype positive individuals. J.J. and A.V. conducted data analysis and interpretation and verified the results. J.J. and G.N.N. wrote the initial draft of the manuscript. A.C., P.K., V.R., P.J., R.D., R.K., S.C., D.B., and A.V. authors provided critical feedback throughout the research process and provided manuscript revision. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors have no conflicts that directly interfere with the contents and implications of this present study. However, there are several disclosures that are associated with the study topic. J.L. reports a consultancy agreement with Viz.ai. R.K. received support from the National Heart, Lung, and Blood Institute of the National Institutes of Health (under award K23HL153775) and the Doris Duke Charitable Foundation (under award, 2022060). In addition to funding from the NIH and the Doris Duke Charitable Foundation, R.K. also receives research support, through Yale, from Bristol-Myers Squibb, Novo Nordisk, and BridgeBio. He is a coinventor of U.S. Pending Patent Applications 63/177,117, 63/428,569, 63/346,610, 63/484,426, 63/508,315, and 63/606,203. He is an academic co-founder of Ensight-AI, Inc. and Evidence2Health, health platforms to improve cardiovascular diagnosis and evidence-based cardiovascular care. D.L.B. discloses the following relationships - Advisory Board: Angiowave, Bayer, Boehringer Ingelheim, Cardax, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, High Enroll, Janssen, Level Ex, McKinsey, Medscape Cardiology, Merck, MyoKardia, NirvaMed, Novo Nordisk, PhaseBio, PLx Pharma, Stasys; Board of Directors: American Heart Association New York City, Angiowave (stock options), Bristol Myers Squibb (stock), DRS.LINQ (stock options), High Enroll (stock); Consultant: Broadview Ventures, Hims; Data Monitoring Committees: Acesion Pharma, Assistance Publique-Hôpitaux de Paris, Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Boston Scientific (Chair, PEITHO trial), Cleveland Clinic, Contego Medical (Chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo; for the ABILITY-DM trial, funded by Concept Medical), Novartis, Population Health Research Institute; Rutgers University (for the NIH-funded MINT Trial); Honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Arnold and Porter law firm (work related to Sanofi/Bristol-Myers Squibb clopidogrel litigation), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), CSL Behring (AHA lecture), Cowen and Company, Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor in Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-Chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Oakstone CME (Course Director, Comprehensive Review of Interventional Cardiology), Piper Sandler, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), WebMD (CME steering committees), Wiley (steering committee); Other: Clinical Cardiology (Deputy Editor); Patent: Sotagliflozin (named on a patent for sotagliflozin assigned to Brigham and Women’s Hospital who assigned to Lexicon; neither I nor Brigham and Women’s Hospital receive any income from this patent); Research Funding: Abbott, Acesion Pharma, Afimmune, Aker Biomarine, Alnylam, Amarin, Amgen, AstraZeneca, Bayer, Beren, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CinCor, Cleerly, CSL Behring, Eisai, Ethicon, Faraday Pharmaceuticals, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Javelin, Lexicon, Lilly, Medtronic, Merck, Moderna, MyoKardia, NirvaMed, Novartis, Novo Nordisk, Otsuka, Owkin, Pfizer, PhaseBio, PLx Pharma, Recardio, Regeneron, Reid Hoffman Foundation, Roche, Sanofi, Stasys, Synaptic, The Medicines Company, Youngene, 89Bio; Royalties: Elsevier (Editor, Braunwald’s Heart Disease); Site Co-Investigator: Abbott, Biotronik, Boston Scientific, CSI, Endotronix, St. Jude Medical (now Abbott), Philips, SpectraWAVE, Svelte, Vascular Solutions; Trustee: American College of Cardiology; Unfunded Research: FlowCo. G.N.N. is affiliated with this journal as an Editor. G.N.N. reports consultancy agreements with AstraZeneca, BioVie, GLG Consulting, Pensieve Health, Reata, Renalytix, Siemens, and Variant Bio; research funding from Goldfinch Bio and Renalytix; honoraria from AstraZeneca, BioVie, Lexicon, and Reata; patents or royalties with Renalytix; owns equity and stock options in Pensieve Health as a cofounder and Renalytix; has received financial compensation as a scientific board member and advisor to Renalytix; serves on the advisory board of Neurona Health; and serves in an advisory or leadership role for Pensieve Health and Renalytix. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, J., Thi Vy, H.M., Charney, A. et al. Multimodal fusion learning for long QT syndrome pathogenic genotypes in a racially diverse population. npj Digit. Med. 7, 226 (2024). https://doi.org/10.1038/s41746-024-01218-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41746-024-01218-1
- Springer Nature Limited