
Abstract
Early patterning of DNA methylation (DNAm) may play an important role in later disease development. To better understand intergenerational epigenetic inheritance, we investigated the correlation between DNAm in blood in mother-newborn and in father-newborn pairs in the Isle of Wight (IoW) birth cohort. For parent-newborn pairs (n = 48), offspring DNAm was measured in cord blood and the parent’s DNAm in whole blood. Mothers’ DNAm was analyzed at birth (Guthrie card), age 18, early and late pregnancy respectively, and fathers’ DNAm was measured during the mother’s pregnancy. Linear regressions were applied to assess the intergenerational correlation of parental DNAm with that of offspring. Among various pairs of mother-newborn and father-newborn DNAm, the pairs where the mothers’ DNAm was measured at age 18 years exhibited the highest number of CpGs with significant intergenerational correlation in DNAm, with 1829 CpGs (0.54%) of the 338,526 CpGs studied (FDR < 0.05). Amongst these 1829 CpGs, 986 (54%) are known quantitative trait loci (QTL) for CpG methylation (methQTL). When the mother’s DNAm was assessed at early pregnancy, the number of CpGs showing intergenerational correlation was the smallest (384 CpGs, 0.11%). The second smallest number of such CpGs (559 CpGs, 0.17%) was found when investigating DNAm in offspring cord blood and father pairs. The low proportions of intergenerationally correlated CpGs suggest that epigenetic inheritance is limited.
Similar content being viewed by others


Introduction
Epigenetic effects are defined as the modification of DNA and its associated gene expression without changes in the genetic code. Early developmental establishment of epigenetic marks may contribute to developmental origins of adult diseases1,2,3. The science of epigenetics has grown considerably in recent years, and it has promise to lead to epigenetic-based treatments or early prevention strategies.
DNAm describes the addition of a methyl group to cysteine in cytosine-phosphate-guanine (CpG) dinucleotides sequences in the DNA. There is little doubt that epigenetics marks such as DNAm are transferred mitotically1. Regarding humans, multiple publications have claimed that epigenetic modifications can actually be transferred from one generation to the next (intergenerational inheritance), thus being meiotically heritable2,3,4,5,6. However, evidence for epigenetic intergenerational inheritance is limited in humans7,8. In particular, erasure of DNAm during early development conflicts with the idea of epigenetic inheritance9,10. It is considered that active demethylation mainly occurs in the paternal genes and that the maternal genes are mainly passively demethylated10,11,12,13,14,15.
Others have suggested that the intergenerational similarity in DNAm is attributable to genetic effects on DNAm such as methylation quantitative trait loci (methQTL)16,17,18, i.e., single nucleotide polymorphisms (SNPs) associated with higher or lower methylation of specific CpGs in cis or trans. A genetic contribution to DNAm correlation between generations is also supported by findings that methylation and gene expression differences are smaller in monozygotic compared to dizygotic twins19,20. CpG sites are not physically situated in a linear space due to the three-dimensional structure of DNA packaging. Therefore, some “distant” CpG sites measured in a two-dimension space are influenced by neighboring methQTLs2 or SNPs within linkage disequilibrium regions. In this situation, epigenetic inheritance due to genetic factors in DNAm of these CpGs may also be observed.
An alternative explanation is that maternal exposures or diseases experienced during pregnancy may produce epigenetic marks in offspring and a higher risk of diseases (indirect inheritance)21. This could then lead to the impression of inter- or intergenerational transmission of DNAm. These exposures or diseases may re-create risk-increasing epigenetic marks in utero, and thus bridge the maternal and offspring DNAm.
A few studies have investigated DNAm among family members at various ages but have not analyzed DNAm at birth2,22. To assess inheritance, Zaghlool et al. investigated trimodality of DNAm in adults of 15 Qatari families with complex pedigree structures2. They predicted that individuals who inherited two unmethylated CpG alleles would display low or zero overall CpG methylation, individuals who inherited one unmethylated and one methylated CpG allele would show a hemi-methylated signal, and individuals who inherited two methylated CpG alleles would show a fully methylated signal. However, the trimodality that they detected was explained by methQTLs. Twin studies in children reported an average genetic heritability of DNAm between 20–34%16,23,24. These studies distinguish between epigenetic heritability and heritability to common effects. However, associations between parents and newborns were not addressed in these twin studies. Hu et al., recently found that there are high correlations between mothers and newborns in whole blood DNAm on both autosomes and chromosome X25.
To improve our understanding of intergenerational epigenetic inheritance, we conducted a study on DNAm in blood samples from mother-newborn and father-newborn pairs in the Isle of Wight birth cohort. To address the different conditions shared during pregnancy by mother and fetus, we also investigated associations between DNAm in offspring cord blood samples with that in mothers sampled at the following time points: late pregnancy, early pregnancy, prepregnancy at age 18 years, and at birth (when the mother was born).
Methods
Isle of Wight F1- and F2-cohorts
The IoW birth cohort study was initiated and conducted at the David Hide Asthma and Allergy Research Centre on the Isle of Wight, UK since 1989/199026. In total, 1,456 children (F1-cohort) born on the Isle of Wight (IOW), from January 1989 to February 1990, and were followed up at 1, 2, 4, 10, 18, and 26 years of age26. In total, 431 offspring of the F1 cohort were recruited between 2010 and 2015 (F2-cohort). We received ethics approval from the Isle of Wight, Portsmouth and SE Hampshire Local Research Ethics Committee. The informed consents were obtained from all subjects in the F1 cohort and the legal guardians of the F2-cohort. The experimental protocols used in the current study were approved by the Isle of Wight, Portsmouth and SE Hampshire Local Research Ethics Committee and at the University of Memphis (IRB ID: 2423).
Sample collection
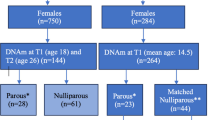
In the F1 generation, DNA was isolated from dried blood spots on Guthrie cards of 796 neonates using a method based on the procedure described by Beyan et al.27. DNA was also extracted from peripheral blood sampled at age 18 years (n = 249), in early (n = 132) and late pregnancy (n = 131) in F1 mothers, and in F1 fathers during the pregnancy of the mothers (n = 96). In the F2 generation, DNA was extracted from 192 cord blood from samples. Figure 1 shows the timeline of the study design when the samples were collected.

Study design of epigenetic inheritance analyses with the timeline for blood sample collection.
For peripheral and cord blood, standard salting out procedure was used to isolate DNA and its concentration was determined by Qubit quantitation. One microgram of DNA was bisulfite-treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, CA, USA), following the manufacturer's standard protocol.
The Infinium HumanMethylation450 BeadChip (450 K array) or EPIC BeadChip V1 (EPIC array) from Illumina (Illumina, San Diego, CA, USA) were used to measure the levels of DNAm at the genome scale, using the manufacturer’s standard protocol (Fig. 2).

Volcano plots of genome-scale regression coefficients in DNA methylation between F2 cord blood and F1 parents at different ages of the mothers (at birth, 18 years, early pregnancy, and late pregnancy) and during pregnancy for fathers.
Preprocessing of DNA methylation
F2 cord DNA methylation data
Two different runs were performed with DNAm measured in 129 samples using 450 K arrays and 63 samples using EPIC arrays. Samples from 450 K and EPIC arrays were pre-processed separately and the data were combined afterwards. Only shared probes between EPIC and 450 K arrays were used in subsequent analyses. The R package ComBat28 was used to remove batch effects in the combined dataset. The CpG sites with probe-SNPs within ten base pairs and with minor allele frequency (MAF) greater than 0.007 (representing ~ 10 subjects in the complete study cohort) were excluded. In total, DNAm at 365,697 CpG cites on autosome chromosomes from 192 samples were analyzed. The same methods for quality control and pre-processing were applied to the DNAm data at different time points in the F1 generation, as discussed below.
F1 parent DNA methylation data
For DNAm from F1 Guthrie cards, quantitative measurements of genome-wide DNAm was performed using the EPIC array. In total, 796 samples with 551,710 autosomal CpGs were included for analysis. The datasets of DNAm consisted of 365,697 CpGs from 249 samples (245 450 K and 4 EPIC) in F1 mothers aged 18 years. For F1 mothers during pregnancy, 372,662 autosomal CpGs from 132 F1 mothers during early pregnancy (129 450 K arrays and 3 EPIC arrays) and 131 during late pregnancy (110 450 K arrays and 21 EPIC arrays). For F1 fathers, 91 samples were assessed using the 450 K array and 5 the EPIC array. In total, 386,440 autosomal CpGs from 96 F1 fathers were included in the analyses.
Cell type proportions
To estimate cell composition for each sample, we used the function estimateCellCount in R-package minfi that is modified from the approach proposed by Houseman et al.29. For cord blood methylation, white blood cell counts were generated using reference panel from Bakulski et al. 201630. For Guthrie cards, mothers at age 18, maternal pregnancy, and father’s DNAm data, cell proportions were estimated using reference panel from Houseman et al.31. For each study, cell type proportions were first estimated separately for the 450 K and EPIC array samples and then combined for subsequent analyses.
Paring up parents and offspring
From the 192 F2 offspring cord blood samples with available DNAm data, we identified 48 F1 mothers with DNAm at age 18 that matched with 48 F2 offspring (i.e., 48 pairs). To avoid concerns of sample size differences in the assessment of association between offspring and parents across different time points, we used the same sample size for all other pairs. It's important to note that not all adult subjects had DNAm data at all ages. To maximize the overlap among all the pairs, we selected 48 subjects at each life stage of mothers (at birth of the mother, in early pregnancy, and in late pregnancy) and 48 fathers paired with 48 F2 cord samples, resulting in a total of five paired study groups. For parents that had multiple children with DNAm available, the child born earlier was included in the study. As depicted in Supplementary Fig. 1, only two F2 cord blood samples were available across all five paired study groups.
Statistical analyses
The demographic characteristics of the complete sample (192 offspring cord blood samples) and the 48 subjects for each offspring-parent pair were summarized for categorical variables with counts and percentages, and continuous variables with mean and standard deviation (SD), respectively. For categorical variables, the one sample z-test for proportions was used to compare the proportion from each group with the complete data set. For continuous variables, the one sample t-test was used to compare the mean of each group with that of complete F2 offspring data set.
For DNAm, beta-values were converted to M-values using base 2 logit-transformation. To adjust for effect of cell type, the methylation of CpGs (M-value) was regressed on the proportion of the cell types. For DNAm from peripheral blood, we regressed M-values on the proportion of CD4 + T cells, NK cells, B cells, monocytes, neutrophils, and eosinophils. The residuals not explained by cell types were estimated for each sample. Similarly, for cord blood cells, we regressed M-values on the proportion of CD4 + T cells, NK cells, B cells, monocytes, granulocytes, and nucleated red blood cells. The residuals not explained by cell types were estimated for each sample. These estimated residuals were used in the following analysis.
To test the associations in cell-type adjusted DNAm between pairs, we treated DNAm from parents as the predictor and offspring DNAm as the response via linear regressions: cell-type adjusted DNAm of offspring = α + β × cell-type adjusted DNAm parent. False discovery rate (FDR) was used to adjust for multiple testing to prevent the inflation of the type I error. CpGs that had FDR adjusted p-values smaller than 0.05 were considered statistically significant and identified as CpG with a potential for heritability. The Bonferroni adjusted alpha is calculated as Bonferroni adjusted alpha = \({1.47\times 10}^{-7}\).
We characterized identified CpGs by checking whether they have been reported as methQTLs. The ARIES methQTL at birth database (cord.ALL.M.tab) was downloaded from http://www.methQTLdb.org32 in April 2019. In total, 73,807 of the 338,526 CpGs in our study were methQTL sites (21.8%).
We also characterized the identified CpGs by their genomic location relative to the genes as previously decribed33. CpG locations were specified by the Illumina Infinium MethylationEPIC v1.0 B4 Manifest File. The categories are: (1) promoter region, 0–1500 bases upstream of the transcriptional start site (TSS); (2) 5' UTR, within the 5' untranslated region, between the TSS and the start codon, ATG; (3) body, between the start and stop codon irrespective of the presence of introns, exons, TSS, or promoters; and (4) 3' UTR, between the stop codon and the transcription termination site, poly A signal. CpGs not belonging to any of the five regions were considered as being in intergenic regions. In terms of relative location of a CpG site to a CpG island, the following four categories were used: (1) shore, 0–2 kb from the CpG island; (2) shelf, 2–4 kb from the CpG island; (3) island, on the CpG island; and (4) CpGs that are not in any of the three categories were considered as “other”.
Ethics declarations
We received ethics approval from both the Isle of Wight, Portsmouth and SE Hampshire Local Research Ethics Committee (Study Title: A study of epigenetic driven immunological changes in the development of Asthma and Allergy in infancy) and the University of Memphis (IRB ID: 2423). The informed consents were obtained from all subjects in the F1 cohort and the legal guardians of the F2-cohort. The experimental protocols used in the current study were approved by the Isle of Wight, Portsmouth and SE Hampshire Local Research Ethics Committee. The research was performed according to the approved protocol and in accordance with the Declaration of Helsinki.
Results
The basic characteristics of the total offspring samples (n = 192), and subsamples for each paired group are summarized in terms of maternal age, paternal age, gestation age, and maternal mother smoking status in Table 1. The results show that the average maternal ages in the paired study subsamples were significantly younger than the average maternal age of the 192 F2 offspring. The reason is that when a mother had multiple children, we included the first-born child in the study to minimize the impact of previous pregnancies on DNAm. The mean paternal age and gestational age are consistent with those in the complete samples, across the five paired study groups. The percentage of maternal smoking in these five paired groups is not statistically different from the percentage of maternal smoking among the 192 F2 children.
A summary of the CpGs showing statistically significant correlation in DNAm between F1 parent and their offspring at birth after controlling FDR of 0.05 (n = 48) are shown in Table 2. Volcano plots of the regression coefficients are presented in Fig. 1. Among the 338,526 CpGs investigated in this study, 1829 CpGs (0.54%) show an intergenerational correlation between F1 mothers at age 18 and F2 at birth. For DNAm of F1 mothers at their birth (Guthrie cards), 925 CpGs show intergenerational correlation, which is only 0.31% of autosomal CpGs. The number of CpGs showing intergenerational correlation between F1 mothers in early and late in pregnancy and F2 at birth are lower, with 384 (0.11%) and 623 (0.18%) CpGs, respectively. The number of CpGs with DNAm showing correlation between F1 fathers and F2 offspring were also smaller compared to F1 mothers at age 18 and F2 at birth with 559 CpGs (0.17% of total) identified. Overall, among all the five parent offspring pairs, for the F1 mothers at age 18 and F2 offspring pairs, the number of CpGs showing correlation is the largest. Among the identified significant CpGs, the majority of them have positive correlations between F1 parent and F2 offspring as shown in Table 2.
Among the 338,526 CpGs examined in our study, 73,807 are methQTL CpG sites as noted in Gaunt et al.32. We found that at 986 methQTL CpGs, methylation levels are significantly associated between F1 mothers at age 18 and F2 cord blood samples, which is about 53.9% of the 1,829 identified significant CpGs (Table 1). A similar percentage (53.4%, 494 out of 925 significantly associated CpGs) are observed for F1 mother at birth and F2 cord pairs. Among the identified CpGs, there are 49.8% methQTLs for mother at late pregnancy with their F2 offspring and 46.2% for father. It is interesting that only 146 CpGs are methQTL (38% of 373 identified CpGs) for mother at early pregnancy and offspring pairs.
Supplementary Fig. 1 shows Manhattan plots of negative log 10 transformed p-values for the five paired samples. We calculated the percentage of identified CpGs out of the total number of CpGs on each chromosome as presented in Supplementary Table 1. The results show that F1 mothers at birth, F1 mothers at age 18, and fathers with F2 offspring pairs have the highest percentage of intergenerational correlated CpGs on chromosome 1, which aligns with the distribution of all CpGs. However, for mothers during both early and late pregnancy, the highest proportion of intergenerational correlated CpGs is found on chromosome 6. Notably, for mothers during late pregnancy, 12.04% of identified significant CpGs are on chromosome 6, which is considerably higher than the 7.19% observed for all CpGs on the same chromosome.
We also investigated the overlap of the identified intergenerational correlated CpGs among different parent and offspring pairs (Table 3 and Supplemental Fig. 3). For example, among the 925 CpGs with DNAm that were significantly correlated between F1 mothers at birth and F2 offspring at birth, 192 of them are also seen among the CpGs identified for the F1 mothers at age 18 and F2 offspring pairs (7.5% of the total number of common significant CpGs between the two paired groups). The identified CpGs found in F1 fathers and their F2 offspring pairs have minimal overlaps with those identified in F1 mothers and F2 offspring pairs, with none of the overlaps exceeding 2%. We also separated the CpGs into those that are methQTLs and those that are not. We then reanalyzed the overlap for each category across the different parent–offspring pairs. The results are shown in Supplementary Tables 2 and 3. Supplementary Fig. 3 shows the diagram of overlapping CpGs, that are methQTLs and those that are not, both between and among all pairs.
In addition, we compared the relative frequency of identified CpGs within each category of gene position (Fig. 3). Comparing to the distribution of all CpGs, for all the four F1 mothers and F2 offspring pair groups, body and intergenic regions had relative higher percentages and 3’ UTR region has relative smaller percentages. F1 fathers and F2 offspring pairs show a slightly different pattern. The distribution of correlated CpGs from F1 father offspring pairs is similar as the distribution of all CpGs.

Manhattan of genome-scale p-values for testing of regression coefficients in DNA methylation between F2 cord blood and F1 parents at different ages of the mothers (at birth, 18 years, early pregnancy, and late pregnancy) and during pregnancy for fathers. CpGs that reached statistical significance are highlighted in green, while the remaining CpGs have alternating colors of blue and orange for different genes. Two horizontal lines indicate cutoff p-values: the blue line represents \({10}^{-5}\) and red line represents the Bonferroni adjusted alpha \({1.47\times 10}^{-7}\).
The relative frequency of the intergenerational correlated CpGs according to their spatial relationship to a CpG island are presented in Fig. 4 for all the five parent–offspring pairs. The proportion of correlated CpGs on islands is smaller across all the paired study groups comparing to all CpGs. The distributions of these CpGs are comparable among shelf, shore and “other” regions for mother and offspring pairs, while the distributions are quite different in father and offspring pairs.

Summary of identified CpGs in each of the five F2 cord blood and F1 parent pairs, mother at birth, mother at age 18, mother in early pregnancy, mother in late pregnancy, and father, within each category of relation to CpG islands. Each percentage was calculated as the number of identified CpGs in the position divided by the total number of significant CpGs for the pair.
Discussion
In this study we examined the correlation of DNAm between parents and their offspring at birth with mothers’ DNAm measured at multiple time points starting from birth and fathers’ DNAm measured during the pregnancy of the mothers. The largest number of highly correlated CpGs (1829 CpGs) was identified in pre-pregnancy mothers, while the respective pool of CpGs was much smaller in mothers during pregnancy, especially during early pregnancy. In addition, we found that the distribution pattern of the significant GpGs are quite different for mother during pregnancy compared to that of all CpGs and other pairs. For example, for mothers during pregnancy, more CpGs are on chromosome 6 comparing to that of all CpGs and F1 pre-pregnancy mother. The observed results could be due to biological and environmental changes during pregnancy. Pregnancy is known to be accompanied by significant hormonal changes that can influence DNAm patterns34,35. In early pregnancy, these hormonal changes are particularly pronounced as the body adjusts to pregnancy, potentially leading to more substantial shifts in DNAm compared to pre-pregnancy levels. In addition, early pregnancy initiates a range of physiological and metabolic changes that support fetal development. These changes, including alterations in blood composition, immune system adjustments, and changes in nutrient processing, could also influence DNAm patterns across the genome. Pregnancy often triggers changes in lifestyle, diet, and even living environment. These changes may also have impact on DNAm36. Overall, the reduction in the number of correlated CpGs during pregnancy suggests that utilizing mother–offspring pairs during pregnancy for studies on DNAm inheritance may not be ideal due to the significant variability introduced by biological and environmental changes during pregnancy.
Turning to the role of fathers, the total number of CpGs with DNAm correlated between father-offspring pair is 599, which is only about one third compared to the1829 CpGs observed in pre-pregnancy mothers. Furthermore, the CpGs identified as heritable in father-offspring pairs showed minimal overlap with those identified in mother–offspring pairs. Such a minimal overlap was also observed in the allocation of these CpGs on genes and their relation to CpG islands on the chromosomes. We postulate a role of allele-specific DNAm that might have played for the difference observed between father and mothers18, but in depth examination is certainly warranted. Additionally, lifestyle factors and environmental exposures have been found to change sperm DNA methylation patterns37,38,39, which subsequently affect DNA methylation patterns in offspring. Paternal genetic effects and paternal-specific gene expression patterns could also initiate the DNA methylation changes in offspring40. It is also possible that DNAm inheritance from fathers to offspring involves different mechanisms compared to that of maternal DNAm inheritance. Of note, offspring DNAm associated with paternal smoking before conception has been demonstrated to be distinct from that observed with maternal smoking34. Further studies are needed to fully understand the specific mechanism of DNAm inheritance from father and mother respectively.
It should be noted that among the CpGs with significant intergenerational correlation in DNAm between parent and offspring, the majority have positive regression coefficients. However, a small number of CpGs exhibit negative correlations. We extracted the CpGs with at least one negative correlation between parent and offspring and summarized them in Supplementary File 3. Notably, three CpGs, cg05945425, cg17334719, and cg2644635, show negative correlations across all five parent–offspring pairs. Additionally, five CpGs, cg12138286, cg06298519, cg21867092, cg01351072, and cg23716690, show negative correlations between mother and offspring, but a positive correlation between father and offspring. The specific mechanisms underlying these negative DNAm associations are not yet clear. Further studies are needed to explore the reasons behind these observations.
The findings presented are based on regression coefficients and a CpG site showing a statistically significant association between parents and newborns. CpGs showing such intergenerational correlation suggest the possibility of DNAm modifications being transmitted from one generation to the next. There are multiple ways by which the parent generation can influence DNAm in offspring (Fig. 5). Conceptually, these can include inheritance of DNA sequence variants, maternal and paternal conditions, and markers of fetal development. In our study, out of the total 1,829 CpGs that have significant association between F1 pre-pregnancy mothers and F2 offspring, 53.9% were identified as methQTL sites. Similar percentages of methQTL sites were observed in other parent–offspring pairs, with the exception of F1 mothers at early pregnancy. This observation is consistent with previous findings that genetic factors plays a significant role in the inheritance of DNAm16,17,18,19,20,40. In addition to genetic factors, other factors, such as life style and environmental exposure may also influence the DNAm in the offspring. The environmental exposures and lifestyle factors unique to each generation can lead to distinct epigenetic modifications that are not necessarily inherited38. Additionally, the reprogramming of the epigenome that occurs during gametogenesis and early embryonic development can erase or modify epigenetic marks, reducing the likelihood of direct inheritance41. Those factors may help to explain what we have observed in the study that the proportion of intergenerationally correlated CpGs is low, which implying that epigenetic inheritance is limited. Overall, the current findings indicate that while some epigenetic marks are heritable, the majority are subject to dynamic regulation and environmental influences rather than stable transmission.

Potential epigenetic pathways from parents to offspring.
In the current study, not all blood samples were analyzed using same platform. Some of the blood samples from parents were analyzed using the Infinium HumanMethylation450 BeadChip and others using the HumanEPIC V1 BeadChip. This variation in platforms might have introduced some bias, despite rigorous quality control35,36. We recognize this as a limitation of our study.
There exist potential concerns of using different pairs across different stages of life, as this design might have led to increased uncertainty. On the other hand, since the inheritance is assessed based on correlations, if there is such an underlying trend of inheritance, the power is likely to be reduced due to the use of different pairs, the patterns should persist. To eliminate the impact of different sample sizes, we used the same sample size across all parent–offspring pair comparisons, with n = 48 for each group. The consistent sample size across comparisons helps ensure that any observed difference in the detection of significant correlated CpGs are not attributable to variations in statistical power between the groups. However, the relatively modest sample sizes may have constrained our ability to detect CpGs with subtle but potentially significant intergenerational correlations. Additionally, the availability of samples varied across the different ages of the mothers, which means that the 48 offspring paired with their mothers at each age, and with their fathers, were not consistent across all study groups. This variability may have introduced noise into the comparison of identified CpGs between different parent–offspring pairs. Comparing to results by Hu et al.25, we observed a lower percentage of correlated DNAm of CpGs between mother and their offspring. In addition to the difference of the study design between these two studies, we recognize it may also be due to a smaller sample size in our study, which leads to a reduced power. Overall, to address these limitations, future studies should consider employing a larger cohort size with DNAm data available for all the study groups, which would enhance statistical power and potentially yield a more comprehensive understanding of epigenetic inheritance.
We examined the correlation of DNAm profiles between parent peripheral blood and offspring cord blood. The unique characteristics of cord blood, primarily influenced by the presence of nucleated red blood cells, differentiate it from peripheral blood. Although we have adjusted for cell type proportions in our analyses, the distinct biological characteristics of cord blood may still influence the methylation patterns observed, potentially affecting the interpretation of intergenerational epigenetic inheritance. Further research is needed to refine the methods for adjusting for different types of cells in future studies, which could involve more sophisticated modeling of cell-type composition or the development of new analytical techniques for comparing heterogeneous sample types.
Another limitation of our study is that we could only explore inheritance between two generations, namely parents and their offspring. As the F2-egg cells are already present in female F1 newborns, we cannot disregard the potential influence of shared factors between F0-grandmothers and F1-mothers that could impact the observed methylation in female F2 offspring. Additionally, this paper focuses on CpGs on autosomes, while CpGs on sex chromosomes are excluded. Future studies investigating the association of DNA methylation of CpGs on sex chromosomes, particularly between mothers and daughters and fathers and sons, will be very interesting.
Overall, our study shows that the correlation of DNAm in various stages of mother-newborn and father-newborn pairs is low. It suggests that the potential epigenetic inheritances between two generations is limited. It is likely that strong confounding or environmental effects exist that limit the detection of substantial intergenerational inheritance in humans.
Data availability
The datasets used in the current study are available from the corresponding author on reasonable request.
References
Delcuve, G. P., Rastegar, M. & Davie, J. R. Epigenetic control. J. Cell Physiol. 219, 243–250 (2009).
Zaghlool, S. B. et al. Mendelian inheritance of trimodal CpG methylation sites suggests distal cis-acting genetic effects. Clin. Epigenet. 8, 124 (2016).
Skinner, M. K. et al. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. Plos One 8, e66318 (2013).
Ushijima, T. et al. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 13, 868–874 (2003).
Waterland, R. A. & Michels, K. B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 27, 363–388 (2007).
Waterland, R. A. Epigenetic mechanisms and gastrointestinal development. J. Pediatr. 149, S137–S142 (2006).
Krauss-Etschmann, S., Meyer, K. F., Dehmel, S. & Hylkema, M. N. Inter- and transgenerational epigenetic inheritance: evidence in asthma and COPD?. Clin. Epigenet. 7, 53 (2015).
Whitelaw, N. C. & Whitelaw, E. Transgenerational epigenetic inheritance in health and disease. Curr. Opin. Genet. Dev. 18, 273–279 (2008).
Hackett, J. A. et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452 (2013).
Leseva, M., Knowles, B. B., Messerschmidt, D. M. & Solter, D. Erase-maintain-establish: Natural reprogramming of the mammalian epigenome. Cold Spring Harb. Symp. Quant. Biol. 80, 155–163 (2015).
Gu, T. P. et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 (2011).
Rougier, N. et al. Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 12, 2108–2113 (1998).
Iqbal, K., Jin, S. G., Pfeifer, G. P. & Szabo, P. E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U S A 108, 3642–3647 (2011).
Guo, F. et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15, 447–458 (2014).
Duan, L., Liu, Y., Wang, J., Liao, J. & Hu, J. The dynamic changes of DNA methylation in primordial germ cell differentiation. Gene 591, 305–312 (2016).
McRae, A. F. et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 15, R73 (2014).
Gertz, J. et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 7, e1002228 (2011).
Shoemaker, R., Deng, J., Wang, W. & Zhang, K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 20, 883–889 (2010).
Loke, Y. J. et al. The peri/postnatal epigenetic twins study (PETS). Twin. Res. Hum. Genet. 16, 13–20 (2013).
Ollikainen, M. et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 19, 4176–4188 (2010).
Arshad, S. H., Karmaus, W., Zhang, H. & Holloway, J. W. Multigenerational cohorts in patients with asthma and allergy. J. Allergy Clin. Immunol. 139, 415–421 (2017).
Tremblay, B. L., Guenard, F., Lamarche, B., Perusse, L. & Vohl, M. C. Familial resemblances in blood leukocyte DNA methylation levels. Epigenetics 11, 831–838 (2016).
Bell, J. T. & Spector, T. D. DNA methylation studies using twins: What are they telling us?. Genome Biol. 13, 172 (2012).
Grundberg, E. et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am. J. Hum. Genet. 93, 876–890 (2013).
Hu, J. et al. Sex differences in the intergenerational link between maternal and neonatal whole blood DNA methylation: A genome-wide analysis in 2 birth cohorts. Clin. Epigenet. 15, 51 (2023).
Arshad, S. H. et al. Cohort profile: The isle of wight whole population birth cohort (IOWBC). Int. J. Epidemiol. 47, 1043–1044i (2018).
Beyan, H. et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 22, 2138–2145 (2012).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Aryee, M. J. et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Bakulski, K. M. et al. DNA methylation of cord blood cell types: Applications for mixed cell birth studies. Epigenetics 11, 354–362 (2016).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
Gaunt, T. R. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17, 61 (2016).
Jiang, Y. et al. Epigenome wide comparison of DNA methylation profile between paired umbilical cord blood and neonatal blood on Guthrie cards. Epigenetics, 1–8 (2019).
Chen, S. et al. Consistency and variability of DNA methylation in women during puberty, young adulthood, and pregnancy. Genet. Epigenet. 9, 1179237X17721540 (2017).
Andrawus, M., Sharvit, L. & Atzmon, G. Epigenetics and pregnancy: Conditional snapshot or rolling event. Int. J. Mol. Sci. 23, 12698 (2022).
Fradin, D. et al. DNA methylation dynamics during pregnancy. Front. Cell Dev. Biol. 11, 1185311 (2023).
Greeson, K. W., Crow, K. M., Edenfield, R. C. & Easley, C. A. IV. Inheritance of paternal lifestyles and exposures through sperm DNA methylation. Nat. Rev. Urol. 20, 356–370 (2023).
Liu, Y. et al. Effects of paternal exposure to cigarette smoke on sperm DNA methylation and long-term metabolic syndrome in offspring. Epigenet. Chromatin 15, 3 (2022).
Wu, D. et al. Future in the past: paternal reprogramming of offspring phenotype and the epigenetic mechanisms. Arch. Toxicol. 98, 1685–1703 (2024).
Zhang, X. & Hocher, B. Parental genetic effects on the offsprings phenotype without direct transmission of the parental gene itself-pathophysiology and clinical evidence. Am. J. Physiol. Cell Physiol. (2024).
Wilkinson, A. L., Zorzan, I. & Rugg-Gunn, P. J. Epigenetic regulation of early human embryo development. Cell Stem Cell 30, 1569–1584 (2023).
Acknowledgements
We thank the High Performance Computing (HPC) facility at the University of Memphis for computation support.
Author information
Authors and Affiliations
Contributions
YJ, HZ and WK conceived the biostatistical design of the study, interpreted the data, and drafted the manuscript. YJ conducted the statistical analysis. SC conducted data cleaning and pre-processing of DNA methylation data. HA and JH supervised the data and sample collection and helped drafting the manuscript. SE conducted the DNA methylation analysis. All authors critically revised the manuscript for important intellectual content. The manuscript has been read and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, Y., Zhang, H., Chen, S. et al. Intergenerational association of DNA methylation between parents and offspring. Sci Rep 14, 19812 (2024). https://doi.org/10.1038/s41598-024-69317-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69317-3
- Springer Nature Limited