
Abstract
Despite advancements made in the therapeutic strategies on hepatocellular carcinoma (HCC), the survival rate of HCC patient is not satisfactory enough. Therefore, there is an urgent need for the valuable prognostic biomarkers in HCC therapy. In this study, we aimed to screen hub genes correlated with prognosis of HCC via multiple databases. 117 HCC-related genes were obtained from the intersection of the four databases. We subsequently identify 10 hub genes (JUN, IL10, CD34, MTOR, PTGS2, PTPRC, SELE, CSF1, APOB, MUC1) from PPI network by Cytoscape software analysis. Significant differential expression of hub genes between HCC tissues and adjacent tissues were observed in UALCAN, HCCDB and HPA databases. These hub genes were significantly associated with immune cell infiltrations and immune checkpoints. The hub genes were correlated with clinical parameters and survival probability of HCC patients. 147 potential targeted therapeutic drugs for HCC were identified through the DGIdb database. These hub genes could be used as novel prognostic biomarkers for HCC therapy.
Similar content being viewed by others
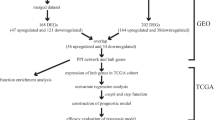


Introduction
Hepatocellular carcinoma (HCC), the second major cause of cancer-associated death worldwide, is a common cancer with poor prognosis due to its high mortality rate and complicated etiology1,2. To some extent, despite advancements made in the therapeutic strategies on HCC, such as surgical resection, transarterial chemoembolization, transplantation and radiofrequency ablation, the survival rate of HCC patient is not satisfactory enough3,4,5,6. Effective treatment interventions for HCC are urgently needed to improve their survival rate and quality of life7,8. Therefore, the identification of novel therapeutic targets and biomarkers will play a vital role in HCC treatment.
Currently, alpha-fetoprotein and des-gamma-carboxyprothrombin have been widely used as prognostic biomarkers in detecting HCC. However, its sensitivity is far from satisfactory9,10. Therefore, it is urgent to identify novel biomarkers for HCC therapy. Several studies have demonstrated that bioinformatics analysis can be used to identify valuable functional genes that could work as prognostic biomarkers11,12,13. Especially, identification of immune-related genes may contribute to HCC therapy. There have been increased immune-related genes in anti-tumour immune responses. For example, several investigators have found that inhibitors of cytotoxic T lymphocyte-associated antigen 4, programmed death-ligand 1 and programed death-1 induce anti-tumoral effects against HCC cells14,15,16. However, only a minority of patients benefits from immunotherapy, emphasizing the need to identify more effective hub genes associated with HCC.
In the current study, we screened out common genes through the intersection of 4 public databases. Then, we performed function enrichment analyses and protein–protein interaction (PPI) network of these genes. We subsequently identified the top ten hub genes by Cytoscape software. Next, we evaluated the correlation between hub genes and prognosis of HCC patients. The potential associations between the hub genes and immune infiltration cells in HCC were also explored. Finally, we obtain potential targeted therapeutic drugs for HCC through DGIdb.
Materials and methods
Ethics statement
Because the current study strictly followed the online database publication guidelines and data access policies, approval from an ethics committee was not required. All methods were performed in accordance with the relevant guidelines and regulations.
Data source
All the data analyzed in this study were derived from public databases. GeneCards is an integrative database that provides information on human genes17. DISEASES, a weekly updated web database, integrates information on gene-disease associations from manually curated literature, cancer mutation data, automatic text mining and genome-wide association studies from existing databases18. Comparative Toxicogenomics Database (CTD), a publicly available database, aims to provide environmental exposure information on gene–disease, chemical–disease and chemical–gene/protein interactions relationships that affects human health19. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and phenotypes, is frequently updated and freely accessible20. HCC-related genes were extracted from GeneCards (https://www.genecards.org/), DISEASES (https://diseases.jensenlab.org/Search), CTD (http://ctdbase.org/) and OMIM (https://www.omim.org/) with the keyword “hepatocellular carcinoma”.
Common gene
The common genes for HCC were obtained by the intersection of the four databases in the Venn diagram online construction website (https://bioinfogp.cnb.csic.es/tools/venny/index.html). All of these common genes were included for further analysis.
Enrichment analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted for the identified common genes by using Metascape (https://metascape.org/)21.
PPI network construction and hub gene identification
The Search Tool for the Retrieval of Interacting Genes (STRING) database (http://string-db.org/) was selected to construct the protein–protein interaction (PPI) network22. The common genes identified previously were uploaded to the STRING database to evaluate the potential PPI relationship. PPI pairs with a combined score more than 0.4 were extracted. Subsequently, the PPI network construction was visualized by Cytoscape software23. Nodes with a high degree tended to be act as an important role in the network. CytoHubba was used to calculate the degree of each node. The top ten genes were then identified as hub genes by the rank of degree.
UALCAN analysis
UALCAN (http://ualcan.path.uab.edu) provides relative transcriptional expression of potential genes between normal and tumor samples as well as association of relative clinical parameters with the transcriptional expression24. In our study, UALCAN was used to perform the mRNA expression of the hub genes in primary HCC tissues and normal control tissues. The relationships between hub genes and clinical parameters were also explored. P < 0.05 (Students t-test) was considered significant.
HCCDB analysis
HCCDB (http://lifeome.net/database/hccdb/home.html), a publicly available web-based database, owns 15 public HCC gene expression datasets to offer a one-stop resource for gene expression analysis in HCC25. We used HCCDB database to validate whether hub genes expression achieved statistical significance in HCC. P < 0.05 was considered significant.
Human protein atlas
The Human Protein Atlas (https://www.proteinatlas.org) is based on immunohistochemistry data of proteins expression26,27. In this study, we obtained immunohistochemistry image for four hub genes from this database.
Kaplan–Meier plotter analysis
The Kaplan–Meier plotter (http://kmplot.com/analysis/) is able to evaluate the effect of hub genes on survival probability in HCC patients. Log rank P-value and hazard ratio (HR, and 95% confidence intervals) were computed28. In this study, the associations between the expression of hub genes and survival state (including overall survival, OS; progression free survival, PFS; recurrence-free survival, RFS; disease free survival, DSS) were analyzed by Kaplan–Meier plotter. We also explored the prognostic value of hub genes in HCC who received sorafenib treatment. Additionally, we demonstrated the prognostic value of combinatory mRNA expression of all ten hub genes in HCC patients and clinical parameters. P < 0.05 was considered significant.
cBioPortal analysis
Multidimensional cancer genomics data sets are available from cBioPortal (http://cbioportal.org)29, we performed all hub genes alterations in the LIHC sample (MSK, Clin Cancer Res 2018; INSERM, Nat Genet 2015; MSK, PLOS One 2018; AMC, Hepatology 2014; RIKEN, Nat Genet 2012; TCGA, Firehose legacy). We explored the genetic alterations of hub genes in per sample by OncoPrint and the prognostic value of these hub genes in OS and DSS of HCC.
TIMER2.0 analysis
TIMER2.0, a comprehensive resource of online server, provides a systematical analysis of immune infiltrates across various cancer types30. In this study, we conducted the associations of hub genes expression with HCC related immune cells, including CD8+ T cells, CD4+ T cells, macrophages, B cells, dendritic cells (DCs) and neutrophils. P < 0.05 was considered significant. Furthermore, we performed the role of hub genes expression combined with macrophage level on OS in HCC patient. The multivariate Cox proportional hazard models of ten hub genes were constructed by adjusted for tumor stage, age, race, gender, macrophage level and tumor purity.
GEPIA
We used GEPIA (http://gepia.cancer-pku.cn/index.html) to analyze the associations between hub genes and immune checkpoints (CD274, CTLA4 and PDCD1)31. Spearman correlation coefficient was used to assess the relationships between hub genes and immune checkpoints expressions in HCC. P < 0.05 was the threshold for significance.
Drug screening
In order to obtain potential targeted therapeutic drugs for HCC, the hub genes were imported into the DGIdb (http://www.dgidb.org)32,33 to acquire potential HCC-associated treatment drugs with the preset filters selected “Approved”.
Results
Identification of common genes
For the purpose of acquiring common genes, we obtained HCC-related genes available in GeneCards, DISEASES, CTD and OMIM databases. After removing the duplicate genes, there were 7816 HCC-related genes in GeneCards, 21283 HCC-related genes in DISEASES, 33724 HCC-related genes in CTD and 505 HCC-related genes in OMIM. Ultimately, 117 common genes were identified by the intersection of the four databases. The Venn Diagram of intersection between all HCC-related genes obtained from these 4 databases was showed in Fig. 1. These 117 genes were listed in Supplementary Table S1.

Venn Diagram of intersection between all the genes of HCC obtained from 4 public databases.
GO and KEGG enrichment analyses of the common genes
Biological processes (BP) analysis indicated that 117 common genes were dramatically enriched in positive regulation of cell death, response to toxic substance, response to lipopolysaccharide and positive regulation of cell adhesion (Fig. 2A). Molecular functions (MF) demonstrated that the common genes were significantly enriched in transcription coregulatory activity, antioxidant activity, kinase binding and R-SMAD binding (Fig. 2B). Cellular components (CC) showed that the common genes were significantly concentrated in membrane raft, perinuclear region of cytoplasm, lysosomal lumen and vesicle lumen (Fig. 2C). Additionally, KEGG analysis revealed that all common genes were mainly enriched in pathways in cancer, proteoglycans in cancer, T cell receptor signaling pathway and TNF signaling pathway (Fig. 2D).

GO, KEGG and PPI network. (A–C) GO enrichment analysis with common genes, (D) KEGG pathway analysis with common genes, (E) PPI network of hub genes.
PPI network construction and hub gene identification
PPI network with 97 nodes and 210 edges were visualized in the Cytoscape. Next, we used the connectivity degree to identify the top ten genes from the PPI network (Fig. 2E). Strong relationship between two genes indicated that their combined score was more than 0.7. Table 1 revealed that jun proto-oncogene (JUN) was the most prominent gene with the highest connectivity degree = 23, followed by interleukin 10 (IL10; degree = 22), CD34 molecule (CD34; degree = 15), mechanistic target of rapamycin kinase(MTOR; degree = 14), prostaglandin-endoperoxide synthase 2 (PTGS2; degree = 13), protein tyrosine phosphatase receptor type c (PTPRC; degree = 13), selectin e (SELE; degree = 12), colony stimulating factor 1 (CSF1; degree = 11), apolipoprotein b (APOB; degree = 10), mucin 1 (MUC1; degree = 10).
Hub gene expression in HCC
The mRNA expression of hub genes in HCC patients was subsequently explored by UALCAN. Among them, JUN (Fig. 3A), IL10 (Fig. 3B), PTGS2 (Fig. 3E), SELE (Fig. 3G), APOB (Fig. 3I) were significantly downregulated in HCC, while CD34 (Fig. 3C), MTOR (Fig. 3D), CSF1 (Fig. 3H) and MUC1 (Fig. 3J) were upregulated. There was no significant difference in the expression of PTPRC (Fig. 3F) between HCC and normal tissues. Furthermore, the mRNA expression of hub genes in HCC, adjacent normal tissue, cirrhotic and healthy samples was acquired from HCCDB database (Fig. 4). JUN was confirmed to be downregulated in HCC tissues compared with adjacent normal tissues in HCCDB1, HCCDB3, HCCDB4, HCCDB6, HCCDB13, HCCDB15, HCCDB17, HCCDB18. The similar results showed for IL10 (HCCDB1, HCCDB3, HCCDB6, HCCDB11, HCCDB13, HCCDB15, HCCDB17, HCCDB18), PTGS2 (from HCCDB1 to HCCDB18), SELE (HCCDB1, HCCDB3, HCCDB4, HCCDB6, HCCDB11, HCCDB13, HCCDB15, HCCDB17, HCCDB18), APOB (HCCDB1, HCCDB3, HCCDB4, HCCDB6, HCCDB12, HCCDB13, HCCDB15, HCCDB17, HCCDB18), PTPRC (HCCDB1, HCCDB3, HCCDB6, HCCDB7, HCCDB12, HCCDB13, HCCDB15, HCCDB16, HCCDB18) and MUC1 (HCCDB11, HCCDB13). However, we found that the expression of CD34 was upregulated in HCCDB1, HCCDB3, HCCDB4, HCCDB6, HCCDB7, HCCDB12, HCCDB13, HCCDB15, HCCDB16 and HCCDB18. The expression of MTOR was upregulated in HCCDB1, HCCDB3, HCCDB4, HCCDB6, HCCDB7, HCCDB13, HCCDB17 and HCCDB18. The expression of CSF1 was upregulated in HCCDB4 and HCCDB17.

mRNA expression of hub genes in HCC tissues and adjacent normal liver tissues. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1. *P < 0.05, **P < 0.01, ***P < 0.001.

The relative expression of hub genes in normal and HCC samples. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1. *P < 0.05, **P < 0.01, ***P < 0.001. Note: Red asterisks represent upregulated genes in HCC patients, and green asterisks represent downregulated genes in HCC patients.
The immunohistochemical images for four genes (IL10, PTGS2, APOB and MUC1) were obtained from the Human Protein Atlas database. These results were consistent with the above results (Fig. 5).

Immunohistochemistry images of four genes in HCC tissues and normal liver tissues. (A) IL10, (B) PTGS2, (C) APOB, (D) MUC1.
Correlations between mRNA expression of hub genes and clinical parameters in HCC patients
For the relationships between mRNA expression of hub genes and clinical parameters in HCC patients were analyzed by UALCAN, such as patients’ individual cancer stages and tumor grades. As was displayed in Fig. 6, mRNA expression of hub genes was significantly associated with patients’ individual cancer stages. The results indicated that the lowest or highest mRNA expression for the vast majority of hub genes was found in patients with advanced cancer stages. The lowest mRNA expression of JUN/PTGS2/PTPRC/SELE/APOB were found in stage 4 (Fig. 6A,E–G,I), while the lowest mRNA expression of IL10 was found in stage 1 (Fig. 6B). The highest mRNA expression of CSF1/MUC1 was found in stage 4 (Fig. 6H,J), while the highest mRNA expression of CD34 and MTOR were found in stage 1 and stage 3, respectively (Fig. 6C,D). Similarly, Fig. 7 demonstrated that the mRNA expression of 8 hub genes was remarkably correlated with tumor grades (Fig. 7A–E,G,I). No significant results were observed for PTPRC and MUC1.

Relationship between mRNA expression of hub genes and individual cancer stages of HCC patients. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1. *P < 0.05, **P < 0.01, ***P < 0.001.

Association of mRNA expression of hub genes with tumor grades of HCC patients. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1. *P < 0.05, **P < 0.01, ***P < 0.001.
We also explored the prognostic value of hub genes in HCC who received sorafenib treatment (Fig. 10). The results demonstrated that high expression of CD34/PTGS2/PTPRC was correlated with favorable OS (Fig. 8A), high expression of JUN/IL10/CD34/PTGS2/SELE was correlated with better RFS (Fig. 8B), high expression of JUN/IL10/CD34/PTGS2/PTPRC/APOB was correlated with better PFS (Fig. 8C), high expression of CD34/PTGS2/PTPRC associated with better DSS (Fig. 8D). Therefore, these hub genes can be used as prognostic indicators for HCC patients who treated by sorafenib.

The prognostic value of ten hub genes in HCC patients who received sorafenib treatment. (A) OS analyses, (B) RFS analyses, (C) PFS analyses, (D) DSS analyses.
In addition, higher combinatory mRNA expression of all ten hub genes was associated with gender, race, alcohol consumption, hepatitis virus, stage, grade, AJCC_T and vascular invasion in HCC (Table 2). These results indicated that combinatory mRNA expression of ten hub genes has a better prognosis in the respective clinical parameters for HCC patients.
Prognostic value of hub genes expression in HCC patients
We then used Kaplan–Meier plotter to perform survival state of ten hub genes in HCC patients. We analyzed the association between combinatory mRNA expression of ten hub genes and prognosis of HCC patients (Fig. 9). Our results revealed that higher combinatory mRNA expression of ten hub genes was remarkably correlated with favorable OS (HR = 0.49, 95% CI 0.34–0.69, P = 3.4E − 06), PFS (HR = 0.67, 95% CI 0.48–0.92, P = 0.012), RFS (HR = 0.62, 95% CI 0.43–0.89, P = 0.008) and DSS (HR = 0.52, 95% CI 0.33–0.82, P = 0.0039) in HCC patients. Next, we found that high expression of CD34/APOB/PTPRC were remarkably correlated with favorable OS of HCC patients (Fig. 10A,E,I), while high expression of MUC1/CSF1 were remarkably correlated with unfavorable OS of HCC patients (Fig. 10M,Q). Similarly, HCC patients with high expression of CD34/APOB/PTPRC/SELE/IL10 were remarkably correlated with better PFS (Fig. 10B,F,J; Supplementary Fig. S1B,F) and RFS (Fig. 10C,G,K; Supplementary Fig. S1C,G). High expression of CD34/APOB/SELE was associated with better DSS (Fig. 10D,H; Supplementary Fig. S1D), nonetheless, the patients with high expression of MUC1 were associated with worse DSS (Fig. 10P). No other significant results were observed in Fig. 10L,N,O,R–T.

Higher combinatory mRNA expressions of all ten hub genes were associated with better prognosis in HCC. (A) OS analysis, (B) PFS analysis, (C) RFS analysis and (D) DSS analysis.

The prognostic value of hub genes in HCC patients. (A–D) CD34, (E–H) APOB, (I–L) PTPRC, (M–P) MUC1, (Q–T) CSF1.
Additionally, we also performed genetic variations of hub genes in 1026 HCC patients using the cBioportal database. The results indicated that genetic variations of hub genes occurred in 203 (20%) of queried HCC patients. Besides, the percentages of genetic variations in hub genes varied from 0.5 to 10% for individual genes (JUN, 0.5%; IL10, 4%; CD34, 4%; MTOR, 3%; PTGS2, 4%; PTPRC, 6%; SELE, 5%; CSF1, 0.7%; APOB, 10%; MUC1, 6%) (Supplementary Fig. S2A). Genetic alteration of hub genes was shown to be associated with worse OS (P = 0.001098) and DSS (P = 0.0202) for HCC patients (Supplementary Fig. S2B,C).
Association between hub genes and immune cell infiltration in HCC
For further understand the relationship between hub genes and immune cell infiltrations in HCC, we used TIMER2.0 to explore their relationship. As showed in Table 3, significant correlations between each of the hub genes and tumor purity were found in HCC tissues. Especially, these ten genes showed remarkable correlations with infiltrating levels of B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophil and DC. These correlation coefficients (COR) were listed from low to high for B cells (0.171–0.375; P < 0.05), CD8+ T cells (0.118–0.396; P < 0.05), CD4+ T cells (0.19–0.278; P < 0.05), macrophages (0.207–0.473; P < 0.05), neutrophil (0.14–0.452; P < 0.05) and DCs (0.138–0.595; P < 0.05). these results demonstrated that hub genes were remarkably associated with tumor-associated B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophil and DCs in the HCC microenvironment.
CD274, PDCD1 and CTLA4 are vital immune checkpoints that are responsible for tumor immune escape. The associations of hub genes with CD274, PDCD1 and CTLA4 were evaluated. Hub genes expression was markedly associated with CD274 (spearman correlation coefficient ranging from 0.16 to 0.61), CTLA4 (spearman correlation coefficient ranging from 0.13 to 0.69) and PDCD1 (spearman correlation coefficient ranging from 0.16 to 0.62) (Supplementary Fig. S3A–J). These findings demonstrated that these hub genes may play an important role in tumor immune escape in HCC patients.
mRNA expression of hub genes with high level of infiltrated macrophages predicted unfavorable OS in HCC
Tumor associated macrophages (TAMs), known as macrophages infiltrating tumors, contribute to tumor initiation, progression, and metastasis. Each of hub genes combined with TAMs was evaluated in prognostic efficiency of HCC patients in this present study. The results were showed in Fig. 11, low expression of CD34 and MUC1 with higher macrophage levels intended to show a worse outcome in HCC (HR = 1.72, P = 0.01 for CD34; HR = 1.60, P = 0.0395 for MUC1). However, under high expression of JUN/IL10/PTGS2/PTPRC/SELE/APOB, higher macrophage levels intended to show a worse outcome in HCC (HR = 1.72, P = 0.0129 for JUN; HR = 1.87, P = 0.0229 for IL10; HR = 1.68, P = 0.0456 for PTGS2; HR = 1.98, P = 0.0118 for PTPRC; HR = 2.07, P = 0.00218 for SELE; HR = 2.61, P = 0.000133 for APOB). Moreover, ten hub genes multivariate Cox proportional hazard models were constructed by adjusted for tumor stage, age, race, gender, macrophage level and tumor purity (Fig. 12). The results showed that, under low expression of IL10, higher macrophage levels intended to have an unfavorable outcome in HCC (HR = 2.36, P = 0.0183). Furthermore, the high level of TAMs could also predict worse prognosis under the high expression of PTPRC (HR = 2.53, P = 0.00901)/SELE (HR = 3.27, P = 0.00145)/APOB (HR = 2.5, P = 0.00617). These results suggested that seven hub genes were independent prognostic biomarker for HCC patients and four hub genes combined with the TAMs would contribute to the prognosis of HCC.

Overall survival analyses for combining the expression of single hub gene with macrophage in HCC patients. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1.

Overall survival analyses for combining the expression of single hub gene with macrophage in HCC patients after adjusting five confounding factors, including age, stage, gender, race, and tumor purity. (A) JUN, (B) IL10, (C) CD34, (D) MTOR, (E) PTGS2, (F) PTPRC, (G) SELE, (H) CSF1, (I) APOB, (J) MUC1.
Drug selection
We screened 147 drugs for interaction with hub genes through the DGIdb database. Among them, 20 targeted JUN, 12 targeted IL10, 1 targeted CD34, 32 targeted MTOR, 53 targeted PTGS2, 10 targeted PTPRC, 4 targeted SELE, 1 targeted CSF1 and 14 targeted APOB. No candidate drugs with interaction with MUC1 were identified (Supplementary Table S2).
Discussion
HCC is frequently diagnosed in advanced stages due to its high heterogeneity34,35. Currently, surgical resection is the first choice for HCC treatment. However, its effectiveness for HCC treatment is still unsatisfactory36,37. Thus, there is an urgent need for identification of novel therapeutic targets and biomarkers in HCC treatment. In our study, multi-databases were comprehensively applied in identifying crucial genes that associated with immune cell infiltrations in HCC. These genes were considered as independent prognostic biomarkers for HCC patients.
A total of 117 common genes were extracted from the intersection of the four databases (GeneCards, DISEASES, CTD and OMIM). The enrichment analysis of BP manifested that these common genes were mainly enriched in positive regulation of cell death, response to toxic substance, response to lipopolysaccharide and positive regulation of cell adhesion. The MF was mainly enriched in transcription coregulatory activity, antioxidant activity, kinase binding and R-SMAD binding. The CC was mainly enriched in membrane raft, perinuclear region of cytoplasm, lysosomal lumen and vesicle lumen. For pathway analysis, the common genes were particularly enriched in pathways in cancer, proteoglycans in cancer, T cell receptor signaling pathway and TNF signaling pathway. These GO terms and KEGG pathway analysis demonstrated that the common genes were enriched in regulating cell function, indicating their close association with tumorigenesis. We then constructed the PPI network based on these genes by STRING, and sub-network of the highest ten degree was identified from the CytoHubba. Next, we explored the mRNA expression of ten hub genes in UALCAN and HCCDB databases, JUN/IL10/PTGS2/PTPRC/SELE/APOB were remarkably downregulated in HCC, while CD34/MTOR/CSF1/MUC1 were upregulated. In addition, we also explored the protein expression of these genes (IL10, PTGS2, APOB and MUC1), the results were consistent with mRNA expression results. There were strong relationships between mRNA expression of hub genes and cancer stages and tumor grades from the results of our study, these hub genes can also be used as prognostic indicators for HCC patients who treated by sorafenib. Meanwhile, combinatory mRNA expression of all ten hub genes was associated with clinical parameters in HCC patients, including gender, race, alcohol consumption, hepatitis virus, stage, grade, AJCC_T and vascular invasion. Furthermore, survival curves analyses by Kaplan–Meier plotter showed that higher combinatory mRNA expression of ten hub genes was correlated with favorable OS, PFS, RFS and DSS in HCC patients. However, genetic alteration of hub genes was shown to be associated with worse OS and DSS for HCC patients. We then explored the prognostic value of single hub genes in HCC patients. Our results suggested that IL10/CD34/PTPRC/SELE/CSF1/APOB/MUC1 had a strong prognostic value for HCC.
These hub genes were confirmed to be strongly associated with infiltrated immune cells in the TIMER2.0 database. These hub genes showed remarkably associated with immune checkpoints (CD274, PDCD1 and CTLA4), which suggested that these hub genes may play an important role in tumor immune escape in HCC patients38. Tumor associated macrophages (TAM), known as macrophages infiltrating tumors, contribute to tumor initiation, progression, and metastasis39,40. Several studies showed that TAM extremely promotes tumor angiogenesis, resulting in a poor prognosis in HCC41,42. The identification of TAM-related genes assists in offering precision therapy for HCC and improving the prognosis of HCC. Thus, we then combined the expression of hub genes with TAM expression to explore the prognostic value in HCC. Under the high expression of JUN/IL10/PTGS2/PTPRC/SELE/APOB, high TAM levels predicted unfavorable prognosis. Meanwhile, under the low expression of CD34/MUC1, high TAM levels predicted unfavorable prognosis. Furthermore, the multivariate Cox regression models demonstrated that IL10/PTPRC/SELE/APOB were independent prognostic biomarker of HCC patients and combined with the TAM would contribute to serving as an important role in clinical prediction of HCC.
IL10, an anti-inflammatory and immunosuppressive factor, is a multifunctional cytokine produced by various immune cells43. It has been showed to modulate cell growth and differentiation. Previous studies suggested that IL10 can activate not only immune cells and immune functions, but also limitation of tumor occurrence and progression under specific microenvironments44,45. IL10 was proved to be an independent predictive survival factor for patients diagnosed with HCC. Various studies suggested that high IL10 expression was correlated with unfavorable prognosis of HCC46,47. However, our study provided evidence against such a conclusion. Results from our study showed that high IL10 expression was correlated with favorable prognosis of HCC. The results were in consistent with the previous studies48,49. Furthermore, after adjustments of tumor stage, age, race, gender and tumor purity, under low expression of IL10, higher macrophage levels intended to show a worse prognosis of HCC. The potential biological function was that TAM inhibits T cell activation and proliferation through IL10 to suppress anti-tumor immunity and promote tumor neovascularization50.
PTPRC, also known as CD45, encodes for a protein and belongs to the protein tyrosine phosphatase (PTP) family. Previous study showed that PTPRC regulates a variety of cellular processes, such as cell growth, differentiation, oncogenic transformation and mitosis51. Recently, several studies have revealed that PTPRC is associated with rheumatoid arthritis52, systemic lupus erythematosus53, Parkinson54, multiple sclerosis55 and T-cell acute lymphoblastic leukemia56. At present, we found that two studies were related to the relationship between PTPRC combined with other genes and HCC in animal experiments57,58. However, there were few relevant clinical studies. Our study suggested that PTPRC combined with the TAM would contribute to acting as an important role in clinical prediction of HCC. Although some possible mechanisms have been proposed, further research is needed59.
SELE, a member of selectin family, usually expressed on activated platelets and endothelial cells, exerts its effects in lymphocytes and monocyte recruitment, rolling, and diapedesis to the inflammatory areas60,61. Previous studies have demonstrated that SELE is associated with coronary artery disease62, coronary heart disease63, coal workers' pneumoconiosis64, hypertension65 and colorectal cancer66. However, there was currently no research on SELE gene associated with HCC. Our study indicated that SELE gene was as a characteristic prognostic biomarker of HCC. Although the underlying this mechanism remains unknown, the mechanism may involve pathways in cancer related to HCC.
APOB, belongs to the apolipoprotein family, forms sub-microscopic spherical particles, which transports dietary lipids from the intestine to the liver via the bloodstream 67. Interestingly, multiple studies have indicated that APOB is associated with non-small cell lung cancer68, gallbladder cancer69, low-grade glioma70 and primary small cell carcinoma of the esophagus71. Study by Lee et al. showed that APOB inactivation is associated with poor outcome in HCC patients72. This was consistent with our findings that HCC patients with high expression of APOB were strongly associated with better overall survival. To date, the underlying mechanism of this relationship remains unknown. However, patients with familial hypobetalipoproteinemia (FHBL) were previously found pathogenic mutations in APOB, which is associated with low-density lipoprotein cholesterol and reducing plasma levels of total cholesterol73. Individuals with FHBL attributable to APOB mutations are intended to hepatic steatosis, liver cirrhosis, and hepatocarcinoma74.
Finally, 147 candidate drugs were found for hub genes through the DGIdb database. The identified drugs could provide reference values for clinical practice when they are validated in vitro studies.
Nevertheless, the present study has several shortcomings. First, further experiments are needed to determine the role of these hub genes in HCC. In addition, although the correlation coefficients between these hub genes and immune cell infiltrations were not absolutely high, the results of this study are credible.
In summary, the ten genes were selected from the PPI network. Most of them were independent prognostic biomarker of HCC patients. Moreover, these genes may exert critical function in HCC progression. In addition, we observed that these genes combined with the TAM would contribute to acting as an important role in clinical prediction of HCC. Overall, these findings suggest that these hub genes may be used as novel prognostic biomarkers for HCC therapy.
Data availability
All the data we obtained are from publicly available databases, the detail information has been described in “Materials and methods”. Further inquiries are available from the corresponding author.
References
McGlynn, K. A., Petrick, J. L. & El-Serag, H. B. Epidemiology of hepatocellular carcinoma. Hepatology 73(Suppl 1), 4–13. https://doi.org/10.1002/hep.31288 (2021).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Disease Primers 7, 6. https://doi.org/10.1038/s41572-020-00240-3 (2021).
Akateh, C. et al. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 25, 3704–3721. https://doi.org/10.3748/wjg.v25.i28.3704 (2019).
Chen, A. et al. Adjuvant transarterial chemoembolization to sorafenib in unresectable hepatocellular carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 36, 302–310. https://doi.org/10.1111/jgh.15180 (2021).
Moeckli, B., Majno, P., Orci, L. A., Peloso, A. & Toso, C. Liver transplantation selection and allocation criteria for hepatocellular carcinoma: A European perspective. Semin. Liver Dis. 41, 172–181. https://doi.org/10.1055/s-0041-1723032 (2021).
Kim, T. H. et al. Proton beam radiotherapy vs. radiofrequency ablation for recurrent hepatocellular carcinoma: A randomized phase III trial. J. Hepatol. 74, 603–612. https://doi.org/10.1016/j.jhep.2020.09.026 (2021).
Deng, Y. et al. Elevated systemic inflammatory responses, factors associated with physical and mental quality of life, and prognosis of hepatocellular carcinoma. Aging 12, 4357–4370. https://doi.org/10.18632/aging.102889 (2020).
Kim, S. et al. Clinical significance of de novo malignancy after liver transplant: A single-center study. Transpl. Proc. 53, 200–206. https://doi.org/10.1016/j.transproceed.2020.02.148 (2021).
Fox, R. et al. Biomarker-based prognosis in hepatocellular carcinoma: Validation and extension of the BALAD model. Br. J. Cancer 110, 2090–2098. https://doi.org/10.1038/bjc.2014.130 (2014).
Abe, T. et al. Glasgow prognostic score and prognosis after hepatectomy for hepatocellular carcinoma. World J. Surg. 41, 1860–1870. https://doi.org/10.1007/s00268-017-3909-7 (2017).
Liu, H. et al. Bioinformatics analysis of differentially expressed rhythm genes in liver hepatocellular carcinoma. Front. Genet. 12, 680528. https://doi.org/10.3389/fgene.2021.680528 (2021).
Zhang, Q. et al. Integrated proteomics and bioinformatics to identify potential prognostic biomarkers in hepatocellular carcinoma. Cancer Manag. Res. 13, 2307–2317. https://doi.org/10.2147/CMAR.S291811 (2021).
Zhang, P. et al. Bioinformatics analysis of candidate genes and pathways related to hepatocellular carcinoma in China: A study based on public databases. Pathol. Oncol. Res. POR 27, 588532. https://doi.org/10.3389/pore.2021.588532 (2021).
Langhans, B. et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol. Immunother. CII 68, 2055–2066. https://doi.org/10.1007/s00262-019-02427-4 (2019).
Wang, J. et al. Hepatocellular carcinoma growth retardation and PD-1 blockade therapy potentiation with synthetic high-density lipoprotein. Nano Lett. 19, 5266–5276. https://doi.org/10.1021/acs.nanolett.9b01717 (2019).
Zhang, Y. et al. Immune-related long noncoding RNA signature for predicting survival and immune checkpoint blockade in hepatocellular carcinoma. J. Cell. Physiol. 235, 9304–9316. https://doi.org/10.1002/jcp.29730 (2020).
Stelzer, G. et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. https://doi.org/10.1186/S12864-016-2722-2 (2016).
Pletscher-Frankild, S., Palleja, A., Tsafou, K., Binder, J. X. & Jensen, L. J. DISEASES: Text mining and data integration of disease-gene associations. Methods 74, 83–89. https://doi.org/10.1016/j.ymeth.2014.11.020 (2015).
Davis, A. P. et al. Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 49, D1138–D1143. https://doi.org/10.1093/nar/gkaa891 (2021).
Amberger, J. S., Bocchini, C. A., Scott, A. F. & Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 47, D1038–D1043. https://doi.org/10.1093/nar/gky1151 (2019).
Zhou, Y. Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. https://doi.org/10.1038/S41467-019-09234-6 (2019).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. https://doi.org/10.1093/nar/gky1131 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. https://doi.org/10.1016/j.neo.2017.05.002 (2017).
Lian, Q. Y. et al. HCCDB: A database of hepatocellular carcinoma expression atlas. Genom. Proteom. Bioinf. 16, 269–275. https://doi.org/10.1016/j.gpb.2018.07.003 (2018).
Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. https://doi.org/10.1126/sciadv.abh2169 (2021).
Sjostedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science https://doi.org/10.1126/science.aay5947 (2020).
Nagy, A., Munkacsy, G. & Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep.-UK. https://doi.org/10.1038/S41598-021-84787-5 (2021).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. https://doi.org/10.1126/scisignal.2004088 (2013).
Li, T. W. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514. https://doi.org/10.1093/nar/gkaa407 (2020).
Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. https://doi.org/10.1093/nar/gkx247 (2017).
Freshour, S. L. et al. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151. https://doi.org/10.1093/nar/gkaa1084 (2021).
Griffith, M. et al. DGIdb: Mining the druggable genome. Nat. Methods 10, 1209–1210. https://doi.org/10.1038/nmeth.2689 (2013).
Friemel, J., Frick, L. & Weber, A. Intratumor heterogeneity in HCC. Aging-US 7, 350–351. https://doi.org/10.18632/Aging.100760 (2015).
Buczak, K. et al. Spatial tissue proteomics quantifies inter- and intratumor heterogeneity in hepatocellular carcinoma (HCC). Mol. Cell Proteom. 17, 810–825. https://doi.org/10.1074/mcp.RA117.000189 (2018).
Abe, T. et al. Glasgow prognostic score and prognosis after hepatectomy for hepatocellular carcinoma (vol 41, pg 1860, 2017). World J. Surg. 41, 1857–1859. https://doi.org/10.1007/s00268-017-3914-x (2017).
Deng, Y. et al. Elevated systemic inflammatory responses, factors associated with physical and mental quality of life, and prognosis of hepatocellular carcinoma. Aging-US 12, 4357–4370. https://doi.org/10.18632/aging.102889 (2020).
Zhang, H. et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. CR 40, 184. https://doi.org/10.1186/s13046-021-01987-7 (2021).
Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. https://doi.org/10.1016/j.cell.2010.03.014 (2010).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416. https://doi.org/10.1038/nrclinonc.2016.217 (2017).
Zhang, J., Chang, L. S., Zhang, X., Zhou, Z. H. & Gao, Y. Q. Meta-analysis of the prognostic and clinical value of tumor-associated macrophages in hepatocellular carcinoma. J. Invest. Surg. 34, 297–306. https://doi.org/10.1080/08941939.2019.1631411 (2021).
Zhao, X. P. et al. Nogo-B fosters HCC progression by enhancing Yap/Taz-mediated tumor-associated macrophages M2 polarization. Exp. Cell Res. https://doi.org/10.1016/J.Yexcr.2020.111979 (2020).
de Waal Malefyt, R., Abrams, J., Bennett, B., Figdor, C. G. & de Vries, J. E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174, 1209–1220. https://doi.org/10.1084/jem.174.5.1209 (1991).
Oft, M. et al. PEGylated IL-10 (pegilodecakin) induces systemic immune activation, CD8+T-cell invigoration and polyclonal T-cell expansion in cancer patients. Cancer Immunol. Res. https://doi.org/10.1158/2326-6074.Cricimteatiaacr18-A016 (2019).
Naing, A. et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J. Clin. Oncol. 34, 3562. https://doi.org/10.1200/Jco.2016.68.1106 (2016).
Qian, Q., Wu, C. P., Chen, J. P. & Wang, W. B. Relationship between IL10 and PD-L1 in liver hepatocellular carcinoma tissue and cell lines. Biomed. Res. Int. https://doi.org/10.1155/2020/8910183 (2020).
Yang, R. N., Gao, N., Chang, Q., Meng, X. C. & Wang, W. H. The role of IDO, IL-10, and TGF-beta in the HCV-associated chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. J. Med. Virol. 91, 265–271. https://doi.org/10.1002/jmv.25083 (2019).
Wang, H., Wang, J., Shi, X. L. & Ding, Y. T. Genetically engineered bone marrow-derived mesenchymal stem cells co-expressing IFN-gamma and IL-10 inhibit hepatocellular carcinoma by modulating MAPK pathway. J. Buon. 22, 1517–1524 (2017).
Shi, Y., Song, Q. W., Hu, D. H., Zhuang, X. H. & Yu, S. C. Tumor-infiltrating lymphocyte activity is enhanced in tumors with low IL-10 production in HBV-induced hepatocellular carcinoma. Biochem. Biophs. Res. Co. 461, 109–114. https://doi.org/10.1016/j.bbrc.2015.03.177 (2015).
Guiducci, C., Vicari, A. P., Sangaletti, S., Trinchieri, G. & Colombo, M. P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Can. Res. 65, 3437–3446. https://doi.org/10.1158/0008-5472.CAN-04-4262 (2005).
Tonks, N. K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846. https://doi.org/10.1038/nrm2039 (2006).
Lee, Y. H. & Bae, S. C. Associations between PTPRC rs10919563 A/G and FCGR2A R131H polymorphisms and responsiveness to TNF blockers in rheumatoid arthritis: A meta-analysis. Rheumatol. Int. 36, 837–844. https://doi.org/10.1007/s00296-016-3476-5 (2016).
Qian, D. et al. JAK2 and PTPRC mRNA expression in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Rheumatol. 39, 443–448. https://doi.org/10.1007/s10067-019-04778-w (2020).
Bottero, V., Santiago, J. A. & Potashkin, J. A. PTPRC expression in blood is downregulated in Parkinson’s and progressive supranuclear palsy disorders. J. Parkinsons Dis. 8, 529–537. https://doi.org/10.3233/JPD-181391 (2018).
Vyshkina, T., Leist, T. P., Shugart, Y. Y. & Kalman, B. CD45 (PTPRC) as a candidate gene in multiple sclerosis. Mult. Scler. 10, 614–617. https://doi.org/10.1191/1352458504ms1115oa (2004).
Porcu, M. et al. Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood 119, 4476–4479. https://doi.org/10.1182/blood-2011-09-379958 (2012).
Zhu, X. et al. Different but synergistic effects of bone marrow-derived VEGFR2(+) and VEGFR2(−)CD45(+) cells during hepatocellular carcinoma progression. Oncol. Lett. 13, 63–68. https://doi.org/10.3892/ol.2016.5411 (2017).
Chen, J. et al. Intratumoral CD45(+)CD71(+) erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. 499, 85–98. https://doi.org/10.1016/j.canlet.2020.12.003 (2021).
Schuette, V. et al. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc. Natl. Acad. Sci. USA 113, 10649–10654. https://doi.org/10.1073/pnas.1605885113 (2016).
Galkina, E. & Ley, K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27, 2292–2301. https://doi.org/10.1161/ATVBAHA.107.149179 (2007).
Fernandez-Borja, M., van Buul, J. D. & Hordijk, P. L. The regulation of leucocyte transendothelial migration by endothelial signalling events. Cardiovasc. Res. 86, 202–210. https://doi.org/10.1093/cvr/cvq003 (2010).
Liao, B. et al. Relationship of SELE A561C and G98T variants with the susceptibility to CAD. Medicine 95, e1255. https://doi.org/10.1097/MD.0000000000001255 (2016).
Dong, Z. Q., Wu, X. J. & Lu, Q. H. Correlations of SELE genetic polymorphisms with risk of coronary heart disease and myocardial infarction: a meta-analysis. Mol. Biol. Rep. 41, 3021–3031. https://doi.org/10.1007/s11033-014-3161-2 (2014).
Wang, T. et al. Polymorphisms in SELE gene and risk of coal workers’ pneumoconiosis in Chinese: A case–control study. PLoS ONE https://doi.org/10.1371/journal.pone.0073254 (2013).
Amabile, N. et al. Increased CD62e(+) endothelial microparticle levels predict poor outcome in pulmonary hypertension patients. J. Heart Lung Transpl. 28, 1081–1086. https://doi.org/10.1016/j.healun.2009.06.005 (2009).
Li, N. et al. SELE gene as a characteristic prognostic biomarker of colorectal cancer. J. Int. Med. Res. 49, 1. https://doi.org/10.1177/03000605211004386 (2021).
Sacks, F. M. The apolipoprotein story. Atheroscler. Suppl. 7, 23–27. https://doi.org/10.1016/j.atherosclerosissup.2006.05.004 (2006).
Deng, W. et al. APOB genotypes and CDH13 haplotypes in the cholesterol-related pathway genes predict non-small cell lung cancer survival. Cancer Epidem. Biomar. 29, 1204–1213. https://doi.org/10.1158/1055-9965.EPI-19-1262 (2020).
Gong, Y., Zhang, L. D., Bie, P. & Wang, H. Z. Roles of ApoB-100 gene polymorphisms and the risks of gallstones and gallbladder cancer: A meta-analysis. PLoS ONE https://doi.org/10.1371/journal.pone.0061456 (2013).
Han, C. et al. Low expression of APOB mRNA or its hypermethylation predicts favorable overall survival in patients with low-grade glioma. Oncotargets Ther. 13, 7243–7255. https://doi.org/10.2147/Ott.S257794 (2020).
Chen, H. et al. ApoB/ApoA-1 ratio as a novel prognostic predictor in patients with primary small cell carcinoma of the esophagus. Front. Oncol. https://doi.org/10.3389/Fonc.2020.00610 (2020).
Lee, G. et al. Clinical significance of APOB inactivation in hepatocellular carcinoma. Cancer Res. https://doi.org/10.1158/1538-7445.AM2018-3404 (2018).
Schonfeld, G. Familial hypobetalipoproteinemia: A review. J. Lipid Res. 44, 878–883. https://doi.org/10.1194/jlr.R300002-JLR200 (2003).
Lonardo, A., Tarugi, P., Ballarini, G. & Bagni, A. Familial heterozygous hypobetalipoproteinemia, extrahepatic primary malignancy, and hepatocellular carcinoma. Dig. Dis. Sci. 43, 2489–2492. https://doi.org/10.1023/a:1026646618643 (1998).
Acknowledgements
The authors are grateful to all subjects who participated in this study.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
Z.L. and X.H. performed the bioinformatics analysis and wrote and revised the manuscript. X.J., N.T. and Y.G. revised the manuscript. L.K. conceived and designed the study, critically revised the manuscript and gave final approval. All authors read and approved the final draft of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, Z., Huang, X., Ji, X. et al. Analysis of multiple databases identifies crucial genes correlated with prognosis of hepatocellular carcinoma. Sci Rep 12, 9002 (2022). https://doi.org/10.1038/s41598-022-13159-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13159-4
- Springer Nature Limited
This article is cited by
-
APOB is a potential prognostic biomarker in hepatocellular carcinoma
Discover Oncology (2024)