
Abstract
We investigated the molecular mechanism(s) by which insulin prevents Bcl2-modifying factor (Bmf)-induced renal proximal tubular cell (RPTC) apoptosis and loss in diabetic mice. Transgenic mice (Tg) mice specifically overexpressing human BMF in RPTCs and non-Tg littermates were studied at 10 to 20 weeks of age. Non-diabetic littermates, diabetic Akita mice +/− insulin implant, Akita Tg mice specifically overexpressing heterogeneous nuclear ribonucleoprotein F (hnRNP F) in their RPTCs and immortalized rat renal proximal tubular cells (IRPTCs) were also studied. BMF-Tg mice exhibited higher systolic blood pressure, urinary albumin/creatinine ratio, RPTC apoptosis and urinary RPTCs than non-Tg mice. Insulin treatment in Akita mice and Akita mice overexpressing hnRNP F suppressed Bmf expression and RPTC apoptosis. In hyperinsulinemic-euglycemic wild type mice, renal Bmf expression was down-regulated with up-regulation of hnRNP F. In vitro, insulin inhibited high glucose-stimulation of Bmf expression, predominantly via p44/42 mitogen-activated protein kinase (MAPK) signaling. Transfection of p44/42 MAPK or hnRNP F small interfering RNA (siRNA) prevented insulin inhibition of Bmf expression. HnRNP F inhibited Bmf transcription via hnRNP F-responsive element in the Bmf promoter. Our results demonstrate that hnRNP F suppression of Bmf transcription is an important mechanism by which insulin protects RPTCs from apoptosis in diabetes.
Similar content being viewed by others

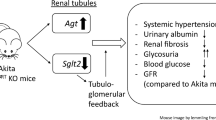
Introduction
Although glomerulopathy is a hallmark of early injury in diabetic kidney disease (DKD), tubulopathy including tubular atrophy and tubulointerstitial fibrosis is a major feature of later stages, closely associated with loss of renal function1,2,3,4. Therefore, tubulopathy is a better predictor of disease progression than glomerular damage5,6,7. Indeed, 71% of glomeruli from proteinuric diabetic patients were found to be attached to atrophic tubules at the glomerulotubular junction, including 8–17% atubular glomeruli (glomeruli without tubular attachment)8,9. The mechanisms underlying tubular atrophy are incompletely understood. One mechanism is apoptosis, which has been implicated in the subsequent loss of various renal cells, including renal proximal tubular cells (RPTCs)10,11,12,13,14,15. Thus, tubular apoptosis may precede tubular atrophy.
Hyperglycemia, hyperlipidemia, oxidative stress and dysfunction of the intrarenal renin-angiotensin system (RAS) have all been implicated in the progression of DKD. We documented that reactive oxygen species (ROS) mediate high glucose (HG) stimulation of angiotensinogen (Agt, the sole precursor of all angiotensins) gene expression in RPTCs in vitro16,17. Hyperglycemia and Agt act in concert to induce hypertension and RPTC apoptosis in type 1 diabetic (T1D) Agt-Tg mice18,19. Conversely, catalase (CAT) overexpression attenuates RPTC apoptosis in T1D CAT-Tg mice20 and T2D db/db CAT-Tg mice21, supporting the view that hyperglycemia via enhanced ROS generation plays a central role in RPTC apoptosis in diabetes.
We used microarray analysis to identify the downstream target genes of ROS, and noted elevated expression of Bcl2 (B-cell lymphoma 2)-modifying factor (Bmf) gene in the RPTCs of db/db mice; the elevated expression was, however, normalized in db/db CAT-Tg mice22. In vitro, Bmf overexpression enhanced and knockdown with small interference RNA (siRNA) decreased RPTC apoptosis in HG milieu. Furthermore, Bmf expression is markedly enhanced and localized to apoptotic RPTCs in human diabetic kidneys22. However, whether overexpression of Bmf would induce RPTC apoptosis and kidney injury in vivo has not been investigated.
Intensive insulin therapy has proven to be the most effective treatment for preventing nephropathy progression in T1D; however, the underlying mechanisms remain incompletely understood23,24. We previously reported that insulin inhibits high-glucose stimulation of renal rat Agt gene expression and RPTC hypertrophy through a putative insulin-responsive element (IRE) in the rat Agt gene promoter that interacts with 2 nuclear proteins, heterogeneous nuclear ribonucleoprotein F and K (hnRNP F/K) in vitro25,26,27. Overexpression of hnRNP F inhibited renal Agt gene expression and attenuated hypertension, kidney hypertrophy and RPTC apoptosis in Akita (T1D) and db/db Tg mice28,29. We further reported that hnRNP F/K mediate insulin inhibition of renal Agt gene expression and that insulin stimulates hnRNP F/K expression through p44/42 MAPK signaling pathway but not through the phosphatidylinositol 3-kinase (PI-3K) pathway in diabetic mice30,31. These findings suggest that insulin inhibition of renal Agt gene transcription and RPTC apoptosis occurs via hnRNP F/K in diabetes.
In the present study, we investigated the impact of Bmf overexpression on RPTC apoptosis in BMF-Tg mice and examined whether hnRNP F mediates, at least in part, insulin regulation of Bmf actions in Akita mice and in rat immortalized RPTCs (IRPTCs) cultured in HG milieu.
Results
RPTC-Specific Expression of Human Bcl2 Modifying Factor (hBMF) Transgene in Transgenic Mice
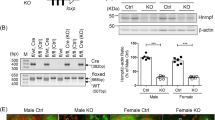
hBMF-Tg mice were generated by inserting myc-hBMF cDNA with the stop codon into a pKAP2 plasmid containing the kidney-specific androgen-regulated protein (KAP) promoter (Fig. 1a). Southern blot analysis revealed the presence of the transgene in heterozygote and homozygote animals from line 148 (Fig. 1b). Tissue-specific analysis by RT-PCR confirmed hBMF mRNA expression in the kidney cortex and RPTs isolated from male hBMF-Tg mice, as well as in the kidney cortex of female hBMF-Tg mice implanted with testosterone pellet but not in other tissues from both male and female mice (Fig. 1c). hBMF transgene was detected in RPTs of male hBMF-Tg mice but not in non-Tg mice and can be differentiated from endogenous mouse Bmf using primers specific for hBMF and mouse Bmf in RT-PCR, respectively (Fig. 1d). WB of isolated RPTs with anti-Bmf or anti-cMyc antibody (Fig. 1e) and immunostaining of kidney sections with an anti-Bmf antibody (that recognizes both human and mouse Bmf) (Fig. 1f) confirmed significantly higher BMF expression in RPTCs from male hBMF-Tg mice than in non-Tg mice. Furthermore, immunofluorescence staining with anti-Bmf and anti-aquaporin-1 (APQ1, a proximal tubule marker) revealed RPTC-specific BMF expression and its co-localization with AQP1-positive RPTCs of male hBMF-Tg mice (Fig. 1g and Supplementary Fig. 1c). These data demonstrate that hBMF expression is RPTC-specific in hBMF-Tg mice.

Generation of hBMF-Tg mice. (a) Schematic map of the kidney androgen-regulated promoter (KAP2)-Myc-hBMF construct. The isolated 17-kb KAP2-myc-hBMF transgene (digested with SpeI and NdeI) was microinjected into 1-cell fertilized mouse embryos obtained from super ovulated C57Bl6 C3H mice. (b) Southern blotting of genomic DNA for founders with biotin-labeled BMF probe. Heterozygous and homozygous F1, F2 and F3 were screened by PCR with specific primers (Table 1). Pc, plasmid positive control. NC, negative control. (c) RT-PCR product showing tissue expression of hBMF mRNA in male and in female Tg mice un-induced or induced with testosterone. hBMF and β-actin fragments are indicated. Female transgenic mice (line #148) mice were induced with placebo pellets or pellets containing 5 mg testosterone with a 21-day release schedule (Cat. #A-121, Innovative Research of America, Sarasota, FL) for 2 weeks prior to RNA isolation. Br, brain; Hr, heart; Lu, lung; Li, liver; Sp, spleen; Ki, kidney; PT, isolated proximal tubule. (d) Specific PCR analysis of hBMF transgene and mouse Bmf in offspring of non-Tg and hBMF-Tg line 148. (e) WB of Bmf and c-Myc protein expression in non-Tg and hBMF-Tg mice. (f) Representative immunostaining for Bmf expression in non-Tg and hBMF-Tg mice. (g) Representative colocalization of immunostaining for Bmf and aquaporin 1 (AQP1) in male non-Tg and hBMF-Tg mouse kidneys (x200). P: proximal tubule, G: glomerulus. Scale bar = 50 µm.
Overexpression of hBMF Increases Systolic Blood Pressure and Kidney Injury in Tg Mice
Longitudinal systolic blood pressure (SBP) measurements revealed consistently higher SBP in hBMF-Tg mice than in non-Tg mice from week 11 to 20 (Fig. 2a). Cross-sectional SBP measurements at week 20 showed significantly elevated SBP (on average by ~8 mm Hg) in hBMF-Tg mice as compared with non-Tg mice (Table 1).

BMF overexpression induces systemic hypertension, glomerulo-tubular fibrosis and pro-fibrotic gene expression in Tg mouse kidneys at week 20. (a) Longitudinal changes in SBP (measured 2 to 3 times per mouse per week in the morning without fasting). Baseline SBP was recorded daily over 5 days before initiation of measurements. (b) Urinary albumin/creatinine ratio (ACR) in non-Tg and hBMF-Tg mice. (c) PAS staining and tubular injury score. (d) Masson’s trichrome staining and semi-quantification of staining. (e) TGF-β1 immunostaining and semi-quantification in non-Tg and hBMF-Tg mice. Scale bar = 50 µm. RT-qPCR of TGF-β1 (f), FN1 (g) and Col Iα (h) mRNA in freshly-isolated RPTs from non-Tg and hBMF-Tg mice. Values are means ± SEM, n = 6. *p < 0.05; **p < 0.01; ***p < 0.005; NS, Not significant.
Marked increases in urinary albumin/creatinine ratio (ACR) were observed in hBMF-Tg mice at 20 weeks of age (Fig. 2b and Table 1). Blood glucose, body weight (BW), kidney weight (KW), tibial length (TL), BW/KW, or BW/TL did not differ significantly between hBMF-Tg mice and non-Tg mice (Table 1). Periodic acid Schiff (PAS) staining of kidney sections revealed mild structural changes in hBMF-Tg compared to non-Tg mice and yielded higher statistically significant tubular injury scores (Fig. 2c). Moreover, more pronounced Masson’s Trichrome staining and TGFβ1 immunostaining were detected in glomerulo-tubular areas of hBMF-Tg mice as compared to non-Tg controls and confirmed by semi-quantification (Fig. 2d,e, respectively). RT-qPCR showed significantly increased mRNA levels of TGFβ1 (Fig. 2f), fibronectin (Fn1) (Fig. 2g) and collagen 1α (Col 1α) (Fig. 2h) in RPTs of hBMF-Tg mice compared to non-Tg mice, indicating that BMF overexpression induces tubulointerstitial fibrosis in kidneys of hBMF-Tg mice. Moreover, morphological measurements revealed significant increases in glomerular tuft volume and RPTC volume in hBMF-Tg mice as compared to non-Tg mice (Table 1).
Overexpression of BMF Stimulates RPTC Apoptosis and Increases Urinary RPTCs
The percentage of TUNEL-positive cells was significantly higher in hBMF-Tg mice as compared to non-Tg mice (Fig. 3a,b). WB analysis showed increases in Bax and cleaved caspase-3 expression (Fig. 3c,d) without detectable changes in Bcl2 and caspase-3 expression in RPT extracts of hBMF-Tg mice as compared to non-Tg mice. Furthermore, co-immunoprecipitation (co-IP) experiments revealed more BMF co-IP with Bcl2 whereas less Bcl2 co-IP with Bax in RPTs of hBMF-Tg mice than non-Tg mice (Fig. 3e). These data would indicate that in hBMF-Tg mice, BMF predominantly binds Bcl2 and dissociates it from Bax, thereby tipping the balance of the Bax/Bcl2 ratio towards caspase-3 activation and subsequently RPTC apoptosis.

Overexpression of BMF induces RPTC apoptosis in mouse kidneys at age 20 weeks. (a) TUNEL (green fluorescence) Assay. Magnification x200. Arrowheads indicate apoptotic cells. P: proximal tubule, G: glomerulus. Scale bar = 50 µm. (b) Semi-quantitation of apoptotic cells in mouse kidneys. (c) WB of Bax, Bcl-2, caspase-3 and cleaved (c)-caspase-3, in freshly-isolated RPTs from non-Tg and hBMF-Tg mice and (d) densitometry analysis. Values are means + SEM, n = 6. **p < 0.01; ***p < 0.005; NS, Not significant. (e) Co-immunoprecipitation of Bcl-2 with Bmf and Bax with Bcl2 in RPT extracts from non-Tg and hBMF-Tg mice. The relative densities of bands of co-IP of Bmf/Bcl2 and Bcl2/Bax were quantified by NIH ImageJ software (http://rsb.info.nih.gov/ij/). (f) Analysis of urinary prominin-1 positive RPTCs in non-Tg and hBMF-Tg mice by flow cytometry. (g) Quantitation of urinary RPTCs/total urinary cells in non-Tg and hBMF-Tg mice. Values are means ± SEM, n = 3. ***p < 0.005; NS, Not significant.
As Bmf is known to enhance anoikis32,33, we investigated whether overexpression of BMF would increase the number of RPTCs in the urine as an indirect indicator of loss of RPTCs. Using flow cytometry, we detected a greater percentage of cells in the urine that stained positive for prominin-1, a proximal tubule marker34 in hBMF-Tg than in non-Tg mice (Fig. 3f,g). These data indicate that overexpression of BMF induces RPTC loss, followed by shedding into the urine in hBMF-Tg mice.
Insulin Inhibits Bmf Expression and RPTC Apoptosis in Akita Mice
We next tested whether insulin reduction of RPTC apoptosis in Akita mice is mediated, at least in part, via suppression of Bmf expression. As anticipated, Bmf expression was higher in RPTs of Akita mice than in WT mice and was inhibited by insulin (Fig. 4a). WB for Bmf protein expression in isolated RPTs confirmed these findings (Fig. 4b,c). Co-immunostaining studies showed Bmf localization to TUNEL-positive RPTCs of Akita mice, which was normalized by insulin treatment (Fig. 4d). The percentage of TUNEL-positive RPTCs was significantly higher in Akita than in WT mice and was normalized by insulin treatment (Fig. 4e). RT-qPCR analysis revealed increases in Bmf (Fig. 4f) and Bax mRNA expression (Fig. 4g), with decreases in Bcl-2 mRNA (Fig. 4h) in Akita mice. These changes in the Akita mice were normalized by insulin treatment with the exception of Bax mRNA. Consistently, the Bax/Bcl-2 mRNA ratio was significantly increased in Akita mice and was reversed by insulin treatment (Fig. 4i).

Insulin effect on Bmf expression and RPTC apoptosis in Akita mice at week 16. (a) Bmf immunostaining (magnification 200x) and semi-quantification of relative Bmf immunostaining by NIH Image J software (http://rsb.info.nih.gov/ij/). (b) Representative WB of Bmf and (c) densitometry analysis in freshly-isolated RPTs from WT, Akita and insulin-treated Akita mice. (d) Co-localization of Bmf expression and TUNEL-positive cells in Akita mouse kidneys. Kidney sections were subjected to TUNEL assay to visualize apoptotic cells (green) and then incubated with anti-Bmf antibody followed by anti-goat AlexaFluor 594 to demonstrate Bmf expression (red). Magnification 600X. Arrows indicate cells that stained positively for TUNEL and Bmf. G, glomerulus; and PT, proximal tubule. (e) Semi-quantitation of apoptotic cells in WT, Akita and insulin-treated Akita mice. RT-qPCR of Bmf (f), Bax (g), Bcl-2 (h) and ratio of Bax/Bcl-2 (i) mRNA in freshly isolated RPTs from WT, Akita and insulin-treated Akita mice. Values are means ± SEM, n = 6. *p < 0.05; **p < 0.01; ***p < 0.005; NS, Not significant.
Insulin Inhibits Bmf Expression Independent of its Glucose Lowering Effect
We previously reported that hyperinsulinemia up-regulated hnRNP F expression in RPTCs independent of its glucose lowering effect31. To investigate whether insulin could also inhibit renal Bmf expression independent of its effect on lowering systemic blood glucose in vivo, hyperinsulinemic-euglycemic clamp experiments were performed on non-diabetic WT mice. Consistent with the previous report31, hyperinsulinemia resulted in increases in hnRNP F immunostaining (Supplementary Fig. 1a) and decreases in Bmf immunostaining (Fig. 5a) and Bmf mRNA (Fig. 5b) as compared with saline infusion. WB of Bmf expression (Fig. 5c) confirmed these observations, indicating that insulin suppression of renal Bmf expression occurs independently of its glucose lowering action.

Renal Bmf expression in hyperinsulinemic-euglycemic mice and in Akita hnRNP F-Tg mice. (a) Representative immunostaining of Bmf (magnification X 200), (b) RT-qPCR of Bmf mRNA expression and (c) representative WB and densitometry of Bmf expression in isolated RPTs from WT mice after 3-h infusion with saline (open bars) or insulin (Ins) + D-glucose (solid black bars). Values are mean ± SEM, n = 8 per group. *p < 0.05; ***p < 0.005; NS, not significant. (d) Representative immunostaining for Bmf (magnification X 200). Semi-quantification of relative Bmf immunostaining in (a,d) was assessed by NIH Image J software (http://rsb.info.nih.gov/ij/), (e) Representative Bmf WB and densitometry analysis, and (f) RT-qPCR of Bmf mRNA expression in isolated RPTs from WT, Akita and Akita hnRNP F-Tg mice at 20 weeks of age. Values are mean ± SEM, n = 8 per group. *p < 0.05; **p < 0.01; NS, not significant.
HnRNP F Overexpression Inhibits Bmf Expression in Akita hnRNP F-Tg Mice
To explore the functional relationship between hnRNP F and Bmf expression in diabetes, we compared Akita hnRNP F-Tg mice to non-Tg mice and Akita mice28. Significant decreases in hnRNP F immunostaining (Supplementary Fig. 1b) and increases in Bmf immunostaining were observed in kidneys of Akita mice as compared with non-Akita WT mice, whereas Bmf immunostaining was reduced in Akita hnRNP F-Tg mice (Fig. 5d). WB of Bmf protein expression (Fig. 5e) and RT-qPCR of Bmf mRNA expression (Fig. 5f) confirmed these observations. These data would imply a role for hnRNP F in mediating insulin suppression of Bmf expression.
Insulin Inhibits Bmf Expression via HnRNP F in IRPTCs
Confirming our previous findings22, we found that HG stimulated Bmf mRNA expression in cultured IRPTCs and that insulin reversed this finding (Fig. 6a). Insulin treatment also prevented the stimulatory effect of HG on rat Bmf promoter (N-1370/+102) activity (Fig. 6b). Pharmacological blockade of p44/42 MAPK (with PD98059 and U0126) effectively prevented insulin inhibition of Bmf promoter activity, whereas blockade of PI-3-Kinase with wortmannin was without effect (Fig. 6b). Moreover, transfection with siRNA of p44 MAPK or p42MAPK also abolished insulin inhibition of Bmf promoter activity in HG, whereas scrambled (Scr) siRNA had no effect (Fig. 6c).

Effect of siRNA of p44/42 MAPK or hnRNP F and hnRNP F cDNA on Bmf gene expression in IRPTCs. (a) Bmf mRNA expression in IRPTCs cultured in NG or HG in the presence or absence of insulin. (b) IRPTCs stably transfected with pGL4.20-rat Bmf gene promoter were incubated in NG or HG DMEM ± insulin for 24 h with or without pharmacological inhibitors or transiently transfected with siRNA of p42 MAPK or p44 MAPK (c) or with siRNA of hnRNP F (d). IRPTCs transiently transfected with pCMV empty or pCMV-hnRNP F plasmid and scrambled (Scr) siRNA or siRNA of hnRNP F to determine hnRNP F mRNA (e) or Bmf mRNA (f) expression. Luciferase activity in cells cultured in NG medium was considered as 100%. The results are expressed as percentage of control (mean ± SEM, n = 3). Each value represents the mean ± SEM (n = 3) assayed in duplicate. **p < 0.01; ***p < 0.005; NS, not significant.
Furthermore, siRNA of hnRNP F, but not Scr siRNA abolished insulin inhibition of Bmf promoter activity in HG (Fig. 6d). Consistently, we detected 10-fold increase and 70% decrease of hnRNP F mRNA expression when IRPTCs were transfected with pCMV-hnRNP F-HA plasmid and siRNA of hnRNP F, respectively (Fig. 6e). In contrast, Bmf mRNA expression decreased by 60% and increased 175% when IRPTCs were transfected with pCMV-hnRNP F-HA and hnRNP F siRNA as compared to controls, respectively (Fig. 6f).
Identification of hnRNP F-Response Element (RE) in Rat Bmf Promoter
To identify putative hnRNP F-RE that mediate the inhibitory action of insulin on Bmf promoter activity, pGL4.20 plasmid containing different lengths of the rat Bmf promoter were transiently transfected into IRPTCs. pGL4.20-Bmf promoter (N-1370/N + 102), pGL4.20-Bmf promoter (N-1260/N + 102), pGL4.20-Bmf promoter (N-1045/N + 102), and pGL4.20-Bmf promoter (N-965/N + 102) displayed a 32-, 26-, 32- and 29-fold increase in activity compared with control pGL4.20 plasmid in IRPTCs in NG (Fig. 7a). Deletion of nucleotides N-1370 to N-365 significantly decreased pGL4.20-Bmf promoter (N-365/+102) activity to just 8-fold higher than plasmid pGL4.20 (Fig. 7a). Insulin prevented HG stimulation on pGL4.20-Bmf promoter (N-1370/N + 102) and pGL4.20-Bmf promoter (N-1260/N + 102) activity whereas insulin failed to affect pGL4.20-Bmf promoter (N-1045/N + 102), pGL4.20-Bmf promoter (N-965/N + 102) and pGL4.20-Bmf promoter (N-365/N + 102) activity (Fig. 7b).

Identification of putative IRE or hnRNP F-RE in the Bmf gene promoter. (a) Luciferase activity of plasmids containing various lengths of Bmf gene promoter in NG medium or (b) HG medium ± insulin in IRPTCs. Luciferase activities were normalized by co-transfecting the vector pRC/RSV containing beta-galactosidase cDNA. Control IRPTCs in NG (open bars), IRPTCs in HG (solid black bars) and IRPTCs treated with insulin in HG (solid light grey). (c) The activity of 1 μg of the full-length Bmf gene promoter ± deletion of putative IRE(N-1086 to N-1081: 5′-AGGGGG-3′), (N-997 to N-991: 5′-GAGGGGC-3′) and (N-402to N-395: 5′-CCCCCGC-3′) in the Bmf gene promoter in IRPTCs in NG or HG medium ± insulin. (d) The activity of 1 μg of the full-length Bmf gene promoter with or without deletion of putative IRE transfected with hnRNP F cDNA in IRPTCs in NG medium. Values are mean ± SEM, n = 3. (*p < 0.05; ***p < 0.005 NS, not significant). (e) EMSA of putative biotinylated IRE (N-1093 to N-1072) with RPTC nuclear proteins with or without excess unlabeled WT IRE or mutated IRE. For supershift rabbit anti-hnRNP F or rabbit IgG was added to the reaction mixture and incubated for 30 minutes on ice before incubation with biotinylated probe. The results are representative of 3 independent experiments. SS, supershift band.
Intriguingly, Bmf promoter with deletion of nucleotides N-1086 to N-1081, pGL4.20-Bmf promoter (N-1370/N + 102 ∆ N-1086/N-1081) completely abolished the response to insulin in HG as compared to pGL4.2-Bmf promoter (N-1370/N + 102) (Fig. 7c). In contrast, Bmf promoter with deletion of N-997 to N-991 and N-402 to N-395 did not alter the response to insulin (Fig. 7c). Furthermore, pGL4.20-Bmf promoter (N-1370/N + 102) activity was significantly suppressed when co-transfected with pCMV-hnRNP F-HA, whereas it had no detectable effect on pGL4.20-Bmf promoter (N-1370/N + 102 ∆ N-1086/N-1081) in NG (Fig. 7d). Moreover, Bmf promoter with deletion of nucleotides N-997 to N-991 and N-402 to N-395 did not affect the inhibitory effect of pCMV-hnRNP F-HA on Bmf promoter activity. These results point toward that nucleotides N-1086 to N-1081 is a putative core RE responding to insulin and hnRNP F.
Indeed, EMSA showed that the WT double-strand DNA fragment (N-1093 to N-1072) binds to IRPTC nuclear proteins, which could be displaced by the WT DNA fragment, but not by mutated DNA fragments (M1, M2 and M3) (Fig. 7e). Moreover, incubation with an anti-hnRNP F antibody, but not with rabbit IgG (control) induced a supershift (SS) of the hnRNP F-RE with nuclear proteins (Fig. 7e).
Discussion
The present study demonstrates that overexpression of hBMF specifically in the RPTCs enhances RPTC apoptosis and loss into the urine in hBMF-Tg mice, indicating a critical role for Bmf in mediating tubular cell apoptosis and loss. Our findings also indicate that insulin treatment prevents RPTC apoptosis and inhibits renal Bmf transcription via a novel putative insulin-response element (IRE) in the Bmf gene promoter that interacts with hnRNP F. These findings identify a novel mechanism by which insulin may prevent nephropathy progression in diabetes.
Bmf is a member of the BH3-only protein family32. Bmf binds to cytoskeletal structures and is sequestered to myosin V motors through association with dynein light chain 2. Certain damage signals, such as the detachment of adherent cells (anoikis) from their substratum trigger the release of Bmf, which through binding to pro-survival Bcl-2 proteins induces dissociation of the Bcl-2/Bax dimer, thereby allowing the pro-apoptotic action of Bax on mitochondria32,33,35,36. Bmf is expressed in various organs, including the kidney33. Mice deficient in Bmf−/−, however, do not display any obvious phenotypic abnormalities37,38. Recent studies by Pfeiffer et al. demonstrated that Bmf not only plays a major role in progressive pancreatic β-cell death in HNF1α-MODY, but also contributes to the pancreatic beta-cell function to maintain glucose homeostasis, independent of cell death signaling39. However, little is known about the role of Bmf in the kidney or in the pathogenesis of diabetes. We previously reported the presence of increased Bmf expression in apoptotic RPTCs in db/db mice as well as in human diabetic kidneys22.
To provide direct evidence for Bmf-mediated RPTC apoptosis in vivo, we generated Tg mice specifically overexpressing hBMF in their RPTCs. Our data demonstrate that overexpression of hBMF in RPTCs indeed induces RPTC apoptosis and loss into the urine in hBMF-Tg mice. Furthermore, overexpression of hBMF increased expression of Bax and cleaved (active) caspase-3 and enhanced Bax binding to Bcl2, consistent with Bax-mediated activation of the intrinsic (mitochondrial) pathway of apoptosis.
The mechanisms underlying increased SBP and ACR in hBMF-Tg mice are incompletely understood. The possibility that up-regulation of TGFβ1 gene expression in RPTCs and its downstream targets Fn1 and Col 1α, leading to higher tubulointerstitial fibrosis that facilitates the development of hypertension has received considerable attention. Indeed, a strong correlation was found between interstitial fibrosis and the development of SBP via a loss or rarefaction of capillaries around the tubules40,41. Our observations of increased glomerular tuft volume, RPTC volume and GFR in hBMF-Tg mice lend additional support to this notion. Furthermore, the observed increases in ACR can be explained, at least in part, by a combination of loss of RPTCs and increased GFR42.
The Akita mouse is an autosomal dominant spontaneous diabetic mouse model (mutation in insulin2) that exhibits many features resembling changes in T1D patients including hypoinsulinemia, hyperglycemia, hypertension and renal dysfunction43,44.
Insulin suppressed Bmf and stimulated hnRNP F expression in RPTCs of hyperinsulinemic-euglycemic mice after 3 h of hyperinsulinemia as compared to Akita mice after 4 weeks of insulin implantation. This rapid transcription is in agreement with previous reports that up-regulated and down-regulated genes in the skeletal muscle and liver were observed within 2 to 4 h under hyperinsulinemic-euglycemic condition45,46. These data demonstrate that insulin impacts RPT Bmf and hnRNP F expression, independent of its glucose-lowering effect.
Our data also revealed significantly higher expression of Bmf protein and mRNA in RPTs of 16 weeks old Akita than WT mice. Overexpression of hnRNP F significantly down-regulated Bmf expression in Akita hnRNP F-Tg mice, suggesting a role for hnRNP F in mediating insulin inhibition of Bmf expression in Akita mice. However, it is noteworthy that we could not exclude the off-target effects of random transgene insertion in mediating insulin suppression of Bmf expression in our hnRNP F-Tg mice. Addressing this issue would require mapping transgene insertion sites which is outside the scope of this present study. Generation of currently unavailable RPTC-specific hnRNP F knockout mice would provide more direct evidence of the role of hnRNP F on Bmf expression. We are now working on developing such a model for future work.
Combining studies with pharmacological inhibitors and siRNAs, we identified roles for the p44/42 MAPK signaling pathway and hnRNP F in mediating insulin suppression of renal Bmf gene transcription. These findings clearly link hnRNP F to mediating insulin inhibition of Bmf gene expression in the diabetic mouse kidney. Nevertheless, additional studies employing RPTC-specific hnRNP F knockout mice are needed to firmly establish this pathway.
The mechanism by which hnRNP F down-regulates renal Bmf gene expression remains to be investigated. A likely mechanism is that hnRNP F suppresses Bmf transcription via binding to a putative IRE in the Bmf promoter. This is supported by the findings that transfection of hnRNP F cDNA considerably decreases Bmf promoter activity, whereas transfection of hnRNP F siRNA reverses insulin effect. DNA sequence analysis revealed 3 GC-rich regions in the Bmf promoter including nucleotides N-1086 to N-1081 (5′-AGGGGG-3′), N-997 to N-991 (5′-GAGGGGC-3′) and N-402 to N-395 (5′-CCCCCGC-3′). Nucleotides N-1086 to N-1081 contain the sequence 5′-AGGGGG-3′ which is homologous to the IRE sequence of N-402 to N-398 (5′-AGGGGG-3′) and N-974 to N-969 (5′-AGGGGG-3′) in the rat Sirtuin-1 and Ace2 promoter, respectively29,47. Deletion of N-1086 to N-1081 in the Bmf promoter markedly reduced insulin- and hnRNP F-down-regulation of Bmf promoter activity in IRPTCs whereas deletion of N-997 to N-991 and N-402 to N-395 had no effect, indicating that the 5′- and 3′-flanking sequences of the core sequence might be important for hnRNP F binding. Moreover, biotinylated IRE (N-1093 to N-1072) binds to nuclear proteins of RPTCs. Addition of anti-hnRNP F antibody yielded a supershift of biotinylated IRE complex with nuclear proteins on EMSA. These findings identify N-1093 to N-1072 as a putative IRE that interacts with hnRNP F and inhibits Bmf transcription.
It is noteworthy that hnRNP F not only regulates Bmf transcription as it also modulates the transcription of Agt28, Sirtuin-129, Nrf231, Ace247 and possibly other genes48. Thus, the hnRNP F effect on Bmf expression might also be mediated by other (indirect) mechanisms. Furthermore, the role of hnRNP F in human pathophysiology remains to be explored. More studies are definitely needed along these lines.
In summary, our results demonstrate that insulin inhibits Bmf transcription and prevents RPTC apoptosis and loss via p44/42 MAPK signaling and hnRNP F expression (Fig. 8) These findings would imply that Bmf activation could aggravate tubulopathy via inducing RPTC loss in the diabetic kidney. However, it remains to be seen whether renal hnRNP F might be a potential target for the prevention of tubulopathy in the diabetic kidney.

Schematic diagram of mechanism of insulin action on inhibition of Bmf gene transcription and tubulopathy. Insulin stimulates p44/42 MAPK signaling and hnRNP F expression. HnRNP F binds to the hnRNP F-RE (N-1086/N-1081) in the Bmf promoter to inhibit Bmf expression and subsequently prevents Bmf-induction of tubulopathy in diabetic kidney.
Methods
Chemical and reagents
D-glucose, D-mannitol, human insulin, PD98059 [a p44/42 MAPK inhibitor] and wortmannin (a PI-3K inhibitor) were procured from Sigma-Aldrich Canada Ltd. (Oakville, ON, Canada). U0126 (an inhibitor of p44/42 MAPK) was purchased from Cell Signaling Technology (New England BioLabs Ltd., Whitby, ON, Canada). Dulbecco’s modified Eagle medium (DMEM, 5 mmol/L D-glucose, catalog no. 12320), penicillin/streptomycin and fetal bovine serum (FBS) were bought from Invitrogen, Inc. (Burlington, ON, Canada). Insulin implants (Linßit, with a release rate of approximately 0.1 unit/implant/day for 30 days) and pGL4.20 [Luc/Puro] vector containing luciferase reporter were obtained from Linshin (Scarborough, ON, Canada) and Promega Corporation (Sunnyvale, CA), respectively. The antibodies used are listed in Supplementary Table 1. Rat genomic DNA was used to clone the Bmf gene promoter N-1370 to N + 102 by conventional polymerase chain reaction (PCR) with specific primers (Supplementary Table 2) and confirmed by DNA sequencing. The Bmf gene promoter then was inserted into pGL4.20 vector via Xho I and Hind III enzyme restriction sites. Scrambled Silencer Negative Control small interfering RNA (siRNA), siRNAs of p44/42 MAPK and hnRNP F were obtained from Ambion, Inc. (Austin, TX). Oligonucleotides and biotin-labeled primers were sourced from Integrated DNA Technologies (Coralville, IA). Kits for LightShift Chemiluminescent electrophoretic mobility shift assay (EMSA) and QuickChange II Site-Directed Mutagenesis were purchased from Agilent Technologies (Santa Clara, CA) and Thermo Scientific (Life Technologies Inc., Burlington, ON, Canada), respectively. Restriction and modifying enzymes were from New England BioLabs, Invitrogen, Roche Biochemicals, Inc. (Dorval, QC, Canada), and GE Healthcare Life Sciences (Baie d’Urfé, QC, Canada).
Generation of human BMF-Tg and akita hnRNP F-Tg mice
Tg mice specifically overexpressing human myc-BMF in their RPTCs were generated using a similar strategy to that described previously20,28,49. In brief, full-length human BMF cDNA fused with Myc-tag was inserted into pKAP2 plasmid at the NotI site. The plasmid pKAP2 containing the KAP promoter that is responsive to androgen was a gift from Dr. Curt D. Sigmund (University of Iowa, Iowa City, IA)50.
Akita hnRNP F-Tg mice were generated by cross-breeding hnRNP F-Tg mice with heterozygous Akita (C57BL/6-Ins2 Akita/J) mice (Jackson Laboratory, Ann Harbor, ME) as previously described28.
Physiological studies
Adult male non-Tg littermates (wild type (WT)) and myc-hBMF-Tg mice were studied at the age of 10 to 20 weeks. Akita mice and Akita hnRNP F-Tg mice were studied at the age of 10 to 16 weeks. Male non-Akita littermates (controls) and Akita mice at the age of 10 weeks treated ± insulin implants and studied from week 12 until week 1641. All animals had access to water and standard mouse chow ad libitum. Animal care and procedures followed the Principles of Laboratory Animal Care [National Institutes of Health (NIH) publication no. 85–23, revised 1985: http://grants1.nih.gov/grants/olaw/references/phspol.htm] and were approved by the Centre de recherche du centre hospitalier de l’Université de Montréal (CRCHUM) Animal Care Committee.
Mouse blood glucose levels after 4 to 5 h of fasting were measured by Accu-Chek Performa (Roche Diagnostics, Laval, QC, Canada). Morning SBP was monitored at least 2–3 times per week in each animal for 6 or 10 weeks with Visitech Systems BP-2000 tail-cuff (Apex, NC)28,30,31,47,51. The mice were acclimatized to the procedure for at least 15–20 min per day for 5 days before the first SBP measurements.
All animals were individually housed in metabolic cages for 8 h during day time for urine collection and blood was collected by cardiac puncture (centrifuged to obtain serum) before euthanasia at 16 or 20 weeks old. Urine samples were assayed for ACR by enzyme-linked immunosorbent assay (ELISA) (Albuwell and Creatinine Companion, Exocell, Inc., Philadelphia, PA). Immediately after euthanasia, the kidneys were removed, decapsulated, and weighed. Left kidneys were processed for histology and immunostaining and right kidneys for RPT isolation by Percoll gradient28,30,31,47,51. Aliquots of freshly isolated RPTs from individual mice were processed for isolation of total RNA and protein.
Fluorescein isothiocyanate-labeled inulin was used to estimate the glomerular filtration rate (GFR) as recommended by the Animal Models of Diabetic Complications Consortium (http://www.diacomp.org/), with slight modifications28,30,31,47,51.
In a separate series of studies, conscious male C57Bl/6 mice (aged 12 to14 weeks) after a 4-hour food restriction were used for hyperinsulinemic-euglycemic clamp experiments as previously described31.
Morphologic studies
Kidney sections of 4 µm thick from five to six animals/group were assessed by standard periodic acid–Schiff (PAS) or Masson’s trichrome staining. Immunohistochemistry staining (IHC) was performed by the standard avidin-biotin-peroxidase complex method (ABC Staining, Santa Cruz Biotechnology, Santa Cruz, CA). Semi-quantitation of the relative staining was done by NIH Image J software (http://rsb.info.nih.gov/ij/).
Tubular injury score, mean glomerular tuft, tubular luminal area, and RPTC volumes were assessed on PAS–stained sections at ×200 magnification as described previously28,30,31,47,51. Briefly, PAS stained images (8–10 fields/kidney) were assessed for tubular injury in the cortex area that displayed tubular dilation, tubular atrophy, cast formation, vacuolization, degeneration, and loss of the brush border. The tubules were evaluated according to the following injury grade (0–3): 0 = no tubular injury, 1 = less than 25% tubules injured, 2 = 25–50% injured, 3 = more than 50% tubules injured52. The score corresponding to tubular injury was calculated for each group by summing and then averaging the grades for each field.
The mean glomerular volume (VG) was determined by the method of Weibel53 with the aid of an image analysis software system (Motics Images Plus 2.0, Motic, Richmond, BC, Canada). The VG was estimated by the mean glomerular tuft area (AT) derived from the light microscopic measurement of 30 random sectional profiles of glomeruli from each group (n = 6 animals per group) using the formula: Vg = β/k x AT1.5, where β = 1.382 (shape coefficient for spheres) and k = 1.1 (size distribution coefficient).
Tubular luminal areas were measured on renal sections (six animals/group; 4 to 5 sections per kidney, 4 random fields per section, 50 tubules around the glomerulus per field) with the same Motics Image Plus 2.0 image analysis software.
Outer cortical RPTs with similar cross-sectional views and clear nuclear structure were selected for cell volume measurement. Mean cell volume from 100 RPTCs was estimated by the Nucleator method54.
Western blotting
Western blotting (WB) was performed as described previously28,30,31,47,51. The relative densities of bands of Bmf, caspase-3, cleaved (active) caspase-3, Bax, Bcl2 and β-actin were quantified by NIH ImageJ software (http://rsb.info.nih.gov/ij/).
Real-Time quantitative PCR
Real-time quantitative (q) PCR was used to quantify the mRNA levels of various genes in RPTs with the forward and reverse primers listed in Supplementary Table 2 28,30,31,47,51.
Cell culture
Rat IRPTCs (passages 13 through 18) were used55. Rat Bmf gene promoter inserted into the plasmid pGL4.20 was stably transfected into IRPTCs as described previously28,30,31,47,51.
To investigate insulin effect, IRPTC stable transformants (75% to 85% confluence) were synchronized for 16 hrs in serum-free DMEM (5 mmol/L D-glucose). Then they were incubated in normal glucose (NG, 5 mmol/L D-glucose plus 20 mmol/L D-mannitol) or HG (25 mmol/L D-glucose) DMEM containing 1% depleted fetal bovine serum (FBS) and insulin (10−7 mol/L or 573 ng/mL) for up to 24 hours ± PD98059 or U0126 or wortmannin31. The cells were then harvested, and the Bmf gene promoter activity was measured by luciferase assay31. IRPTCs stably transfected with pGL4.20 served as controls. In addition, scrambled siRNA or siRNAs of p44/42 MAPK or hnRNP F were transfected into IRPTC stable transformants and cultured for 24 hours, their effects on Bmf gene promoter activity and mRNA expression were assessed.
Flow cytometry analysis of urinary cells
Urine was collected for 6 hrs from individual non-Tg or myc-hBMF-Tg mice placed individually in metabolic cages. Urines from 5 mice in each group were collected, pooled and centrifuged at 500 × g for 10 minutes to pellet down the cells in the urine. Pellets were rinsed with PBS and passed through a 35 μm cell strainer (#352235, Corning Inc., Corning, USA). Each cell suspension was then blocked with 2% FBS in PBS for 10 minutes then stained with DRAQ7 and anti-prominin-1 antibody conjugated with PE-Vio770 (MiltenyiBiotec, BergischGladbach, Germany, 1:25 dilution)34 for 30 minutes at room temperature in the dark, then washed with PBS; equal volumes of suspension from non-Tg or myc-hBMF-Tg mice were subjected to flow cytometry (LSR-II, BD Biosciences). Staining with DRAQ7 was used to assess viability. The data was analyzed using FlowJo V10 (FlowJo, LLC, Ashland, USA).
Statistical analysis
Data are expressed as means ± SEM. Statistical analysis was performed with the Student t test or one-way ANOVA and the Bonferroni test, as appropriate (GraphPad Prism 5.0 software, http://www.graphpad.com/prism/Prism.htm). P < 0.05 was considered to be statistically significant for all tests. (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; NS, non-significant)
References
Drummond, K. & Mauer, M. The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes 51, 1580–1587 (2002).
Schainuck, L. I., Striker, G. E., Cutler, R. E. & Benditt, E. P. Structural-functional correlations in renal disease. II. The correlations. Human pathology 1, 631–641 (1970).
Gilbert, R. E. & Cooper, M. E. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney international 56, 1627–1637, https://doi.org/10.1046/j.1523-1755.1999.00721.x (1999).
Gilbert, R. E. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes 66, 791–800, https://doi.org/10.2337/db16-0796 (2017).
Javaid, B., Olson, J. L. & Meyer, T. W. Glomerular injury and tubular loss in adriamycin nephrosis. Journal of the American Society of Nephrology: JASN 12, 1391–1400 (2001).
Marcussen, N. Tubulointerstitial damage leads to atubular glomeruli: significance and possible role in progression. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 15(Suppl 6), 74–75 (2000).
Lindop, G. B., Gibson, I. W., Downie, T. T., Vass, D. & Cohen, E. P. The glomerulo-tubular junction: a target in renal diseases. The Journal of pathology 197, 1–3, https://doi.org/10.1002/path.1087 (2002).
Najafian, B., Kim, Y., Crosson, J. T. & Mauer, M. Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy. Journal of the American Society of Nephrology: JASN 14, 908–917 (2003).
Najafian, B., Crosson, J. T., Kim, Y. & Mauer, M. Glomerulotubular junction abnormalities are associated with proteinuria in type 1 diabetes. Journal of the American Society of Nephrology: JASN 17, S53–60, https://doi.org/10.1681/asn.2005121342 (2006).
Nagata, S. Apoptosis by death factor. Cell 88, 355–365 (1997).
Kang, B. P. et al. High glucose promotes mesangial cell apoptosis by oxidant-dependent mechanism. American journal of physiology. Renal physiology 284, F455–466, https://doi.org/10.1152/ajprenal.00137.2002 (2003).
Allen, D. A., Harwood, S., Varagunam, M., Raftery, M. J. & Yaqoob, M. M. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 17, 908–910, https://doi.org/10.1096/fj.02-0130fje (2003).
Kumar, D., Zimpelmann, J., Robertson, S. & Burns, K. D. Tubular and interstitial cell apoptosis in the streptozotocin-diabetic rat kidney. Nephron. Experimental nephrology 96, e77–88, https://doi.org/10.1159/000076749 (2004).
Kumar, D., Robertson, S. & Burns, K. D. Evidence of apoptosis in human diabetic kidney. Molecular and cellular biochemistry 259, 67–70 (2004).
Mishra, R., Emancipator, S. N., Kern, T. & Simonson, M. S. High glucose evokes an intrinsic proapoptotic signaling pathway in mesangial cells. Kidney international 67, 82–93, https://doi.org/10.1111/j.1523-1755.2005.00058.x (2005).
Hsieh, T. J. et al. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells. Endocrinology 144, 4338–4349, https://doi.org/10.1210/en.2003-0220 (2003).
Hsieh, T. J. et al. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology 143, 2975–2985, https://doi.org/10.1210/endo.143.8.8931 (2002).
Liu, F. et al. Overexpression of angiotensinogen increases tubular apoptosis in diabetes. Journal of the American Society of Nephrology: JASN 19, 269–280, https://doi.org/10.1681/ASN.2007010074 (2008).
Lo, C. S. et al. Dual RAS blockade normalizes angiotensin-converting enzyme-2 expression and prevents hypertension and tubular apoptosis in Akita angiotensinogen-transgenic mice. American journal of physiology. Renal physiology 302, F840–852, https://doi.org/10.1152/ajprenal.00340.2011 (2012).
Brezniceanu, M. L. et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney international 71, 912–923, https://doi.org/10.1038/sj.ki.5002188 (2007).
Brezniceanu, M. L. et al. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 57, 451–459, https://doi.org/10.2337/db07-0013 (2008).
Lau, G. J. et al. Bcl-2-modifying factor induces renal proximal tubular cell apoptosis in diabetic mice. Diabetes 61, 474–484, https://doi.org/10.2337/db11-0141 (2012).
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin dependent diabetic mellitus. N Engl J Med 329, 977–986 (1993).
de Boer, I. H. et al. Insulin therapy, hyperglycemia, and hypertension in type 1 diabetes mellitus. Arch Intern Med 168, 1867–1873 (2008).
Chen, X. et al. Characterization of a putative insulin-responsive element and its binding protein(s) in rat angiotensinogen gene promoter: regulation by glucose and insulin. Endocrinology 142, 2577–2585 (2001).
Wei, C. C., Guo, D. F., Zhang, S. L., Ingelfinger, J. R. & Chan, J. S. Heterogenous nuclear ribonucleoprotein F modulates angiotensinogen gene expression in rat kidney proximal tubular cells. J Am Soc Nephrol 16, 616–628 (2005).
Wei, C. C. et al. Heterogeneous nuclear ribonucleoprotein K modulates angiotensinogen gene expression in kidney cells. J Biol Chem 281, 25344–25355 (2006).
Lo, C. S. et al. Heterogeneous nuclear ribonucleoprotein F suppresses angiotensinogen gene expression and attenuates hypertension and kidney injury in diabetic mice. Diabetes 61, 2597–2608, https://doi.org/10.2337/db11-1349 (2012).
Lo, C. S. et al. Heterogeneous Nuclear Ribonucleoprotein F Stimulates Sirtuin-1 Gene Expression and Attenuates Nephropathy Progression in Diabetic Mice. Diabetes 66, 1964–1978, https://doi.org/10.2337/db16-1588 (2017).
Abdo, S. et al. Heterogeneous nuclear ribonucleoproteins F and K mediate insulin inhibition of renal angiotensinogen gene expression and prevention of hypertension and kidney injury in diabetic mice. Diabetologia 56, 1649–1660, https://doi.org/10.1007/s00125-013-2910-4 (2013).
Ghosh, A. et al. Insulin Inhibits Nrf2 Gene Expression via Heterogeneous Nuclear Ribonucleoprotein F/K in Diabetic Mice. Endocrinology 158, 903–919, https://doi.org/10.1210/en.2016-1576 (2017).
Puthalakath, H. & Strasser, A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell death and differentiation 9, 505–512, https://doi.org/10.1038/sj/cdd/4400998 (2002).
Puthalakath, H. et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science (New York, N.Y.) 293, 1829–1832, https://doi.org/10.1126/science.1062257 (2001).
Legouis, D. et al. Ex vivo analysis of renal proximal tubular cells. BMC cell biology 16, 12, https://doi.org/10.1186/s12860-015-0058-4 (2015).
Morales, A. A. et al. Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia 18, 41–47, https://doi.org/10.1038/sj.leu.2403183 (2004).
Day, C. L. et al. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. The Biochemical journal 377, 597–605, https://doi.org/10.1042/bj20031251 (2004).
Labi, V. et al. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. The Journal of experimental medicine 205, 641–655, https://doi.org/10.1084/jem.20071658 (2008).
Hubner, A., Cavanagh-Kyros, J., Rincon, M., Flavell, R. A. & Davis, R. J. Functional cooperation of the proapoptotic Bcl2 family proteins Bmf and Bim in vivo. Molecular and cellular biology 30, 98–105, https://doi.org/10.1128/mcb.01155-09 (2010).
Pfeiffer, S., Halang, L., Düssmann, H., Byrne, M. M. & Prehn, J. H. M. BH3-Only protein bmf is required for the maintenance of glucose homeostasis in an in vivo model of HNF1α-MODY diabetes. Cell Death Discovery 1, 15041, https://doi.org/10.1038/cddiscovery.2015.41 (2015).
Grgic, I. et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney international 82, 172–183, https://doi.org/10.1038/ki.2012.20 (2012).
Afsar, B. et al. Capillary rarefaction from the kidney point of view. Clinical kidney journal 11, 295–301, https://doi.org/10.1093/ckj/sfx133 (2018).
Schelling, J. R. Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatric nephrology (Berlin, Germany) 31, 693–706, https://doi.org/10.1007/s00467-015-3169-4 (2016).
Wang, J. et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. The Journal of clinical investigation 103, 27–37, https://doi.org/10.1172/jci4431 (1999).
Yoshioka, M., Kayo, T., Ikeda, T. & Koizumi, A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46, 887–894 (1997).
Coletta, D. K. et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. American journal of physiology. Endocrinology and metabolism 294, E910–917, https://doi.org/10.1152/ajpendo.00607.2007 (2008).
Korsheninnikova, E. et al. Dynamics of insulin signalling in liver during hyperinsulinemic euglycaemic clamp conditions in vivo and the effects of high-fat feeding in male mice. Archives of physiology and biochemistry 113, 173–185, https://doi.org/10.1080/13813450701669084 (2007).
Lo, C. S. et al. Overexpression of heterogeneous nuclear ribonucleoprotein F stimulates renal Ace-2 gene expression and prevents TGF-beta1-induced kidney injury in a mouse model of diabetes. Diabetologia 58, 2443–2454, https://doi.org/10.1007/s00125-015-3700-y (2015).
Wang, E. et al. Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PloS one 7, e51266, https://doi.org/10.1371/journal.pone.0051266 (2012).
Sachetelli, S. et al. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney international 69, 1016–1023, https://doi.org/10.1038/sj.ki.5000210 (2006).
Ding, Y. & Sigmund, C. D. Androgen-dependent regulation of human angiotensinogen expression in KAP-hAGT transgenic mice. American journal of physiology. Renal physiology 280, F54–60 (2001).
Abdo, S. et al. Catalase overexpression prevents nuclear factor erythroid 2-related factor 2 stimulation of renal angiotensinogen gene expression. hypertension, and kidney injury in diabetic mice. Diabetes 63, 3483–3496, https://doi.org/10.2337/db13-1830 (2014).
Véniant, M., Heudes, D., Clozel, J. P., Bruneval, P. & Ménard, J. Calcium blockade versus ACE inhibition in clipped and unclipped kidneys of 2K-1C rats. Kidney Int 46, 421–429 (1994).
Weibel, E. R. Numerical density: shape and size of particles. In Sterological Methods. Vol. 2, Theorectical Foundations. Weiel, E. R., ed. p149–152 (London, Academic Press, 1980).
Gundersen, H. J. G. The nucleator. J Microsc 151, 3–21 (1988).
Tang, S. S. et al. Temperature-sensitive SV40 immortalized rat proximal tubule cell line has functional renin-angiotensin system. The American journal of physiology 268, F435–446 (1995).
Acknowledgements
This manuscript or any significant part of it is not under consideration for publication elsewhere. Parts of data were, however, presented at the 2017 American Society of Nephrology meeting, New Orleans, LA Oct.31-November 5th, 2017. This work was supported by research grants from Canadian Institutes of Health Research (MOP-84363 and MOP-106688 to JSDC, MOP-86450 to SLZ, and MOP-97742 to JGF), Canadian Diabetes Association (NOD_OG-3-14-4472-JC to JSDC), Kidney Foundation of Canada (KFOC 170006 to JSDC) and National Institutes of Health (NIH) of USA (HL-48455 to JRI).
Author information
Authors and Affiliations
Contributions
J.S.D.C. is the guarantor of this work, has full access to all study data, and takes responsibility for data integrity and the accuracy of data analysis. J.S.D.C. contributed to study conception and design, drafted and reviewed/edited the final manuscript. A.G. wrote the first draft of the manuscript and contributed to the discussion. A.G., S.Z., C.S.L., I.C., H.M., M.A.L., S.A. and S.L.Z. contributed to research data. S.L.Z., J.G.F. and J.R.I. contributed to discussion and reviewed/edited the manuscript. All authors gave final manuscript approval. The assistance from CRCHUM Rodent Metabolic Phenotyping core facility for the hyperinsulinemic-euglycemic clamp studies is greatly appreciated.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghosh, A., Zhao, S., Lo, CS. et al. Heterogeneous Nuclear Ribonucleoprotein F Mediates Insulin Inhibition of Bcl2-Modifying Factor Expression and Tubulopathy in Diabetic Kidney. Sci Rep 9, 6687 (2019). https://doi.org/10.1038/s41598-019-43218-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43218-2
- Springer Nature Limited