
Abstract
Genetic linkage maps are an important tool in genetic and genomic research. In this study, two hawthorn cultivars, Qiujinxing and Damianqiu, and 107 progenies from a cross between them were used for constructing a high-density genetic linkage map using the 2b-restriction site-associated DNA (2b-RAD) sequencing method, as well as for mapping quantitative trait loci (QTL) for flavonoid content. In total, 206,411,693 single-end reads were obtained, with an average sequencing depth of 57× in the parents and 23× in the progeny. After quality trimming, 117,896 high-quality 2b-RAD tags were retained, of which 42,279 were polymorphic; of these, 12,951 markers were used for constructing the genetic linkage map. The map contained 17 linkage groups and 3,894 markers, with a total map length of 1,551.97 cM and an average marker interval of 0.40 cM. QTL mapping identified 21 QTLs associated with flavonoid content in 10 linkage groups, which explained 16.30–59.00% of the variance. This is the first high-density linkage map for hawthorn, which will serve as a basis for fine-scale QTL mapping and marker-assisted selection of important traits in hawthorn germplasm and will facilitate chromosome assignment for hawthorn whole-genome assemblies in the future.
Similar content being viewed by others


Introduction
Hawthorn (Crataegus pinnatifida Bunge) belongs to the family Rosaceae and is a widespread fruit tree in China. The fruits are used for food and medicinal purposes. The leaves, fruits, roots, and twigs of hawthorn contain many nutrients, including proteins, fats, dietary fibre, vitamins, flavones, and many minerals. Many studies have focused on hawthorn flavones1,2,3,4,5,6, which are the primary bioactive components7. Flavones are polyphenol secondary metabolites that have low molecular weight and are common in plants; they are produced in response to environmental stress and play a role in defence against predators and pathogens8. Flavones have antioxidant and anticancer properties and can scavenge free radicals9, so their properties have been widely researched for use in agriculture, the chemical industry and medicine. Flavone production is under genetic regulation, but the regulation of flavones in hawthorn has been little studied.
Genetic linkage maps, particularly high-density maps, are one of the most valuable tools for high-throughput selection of superior traits from plant and animal germplasms. Our lab published the first hawthorn genetic linkage map that was constructed using sequence-related amplified polymorphism markers10, but its application value was limited because it included relatively few markers with long marker intervals.
As high-throughput technology and next-generation sequencing (NGS) methods have been developed, many can now quickly genotype thousands of markers in a single step11. Restriction-site associated DNA sequencing (RAD-seq)12, 13, specific length amplified fragment (SLAF) sequencing14, and genotyping by sequencing (GBS, or NGS)15 are powerful tools for constructing high-density genetic linkage maps. For example, Pfender et al.16, Chutimanitsakun et al.17, and Wang et al.18 used RAD-seq to construct high-density genetic linkage maps for ryegrass, barley, and grape, respectively. Poland et al.15 constructed high-density genetic linkage maps for barley and wheat, and Zhang et al.19 constructed a map for jujube based on GBS technology. Several other studies have used SLAF to construct high-density genetic linkage maps for soybean20, sesame21, and Salvia miltiorrhiza Bunge22.
Type IIB endonuclease RAD (2b-RAD) uses restriction enzymes such as BsaXI or AlfI (both insensitive to methylation) to produce uniform tags. This is a simple and flexible method for genome-wide genotyping23.
In this study, our aim was to construct a high-density genetic linkage map for hawthorn using the 2b-RAD method and then conduct fine-scale QTL mapping for flavonoid content in hawthorn leaves. This linkage map will be a powerful tool for research involving both fine-scale QTL mapping and marker-assisted selection of important economic traits of hawthorn germplasm. It will facilitate chromosome assignment for a future whole-genome assembly.
Results
2b-RAD sequencing and markers selection
The Hiseq2500 v2 platform was used to conduct single-end sequencing of 2b-RAD libraries for the parents and 107 progenies. A total of 526,949,839 reads were recovered, including 11,265,798 reads from the seed parent, 12,977,353 reads from the pollen parent, and 502,706,688 reads from the progeny. The sequencing depth was 62× for the seed parent, 52× for the pollen parent, and 23× for the 107 progenies. The sequencing depth detail of the 107 progenies is shown in Fig. 1. After low-quality reads were trimmed, 82.39% of reads from the seed parent, 82.67% of reads from the pollen parent, and 84.50% of reads from the progeny were retained for analysis (Table 1). Using SOAP software24, 117,896 unique tags were generated, including 13,937 dominant tags and 103,959 codominant tags. RAD typing v1.0 software25 was used to genotype the reads, with 47,923 SNPs that were polymorphic markers, including 9,237 dominant markers and 38,686 codominant markers. A Mendelian fit test and genotyping percentage were used to trim SNP markers for 12,951 markers that fit Mendelian ratios (P ≥ 0.05) and possessed a high genotyping rate (the available genotype was found in over 80% of progeny), and the types of all these markers are shown in Fig. 2. These markers were used for constructing parental maps. Finally, 6,390 markers were used for constructing a seed parent map and 7,384 markers were used for constructing a pollen parent map. In total, 823 markers were shared by the two parents (Table 2).

Sequencing depth of 107 progeny. The x-axes indicate average depth and the y-axes indicate individual plant accessions.

Number of markers for each segregation pattern. The x-axes means marker type and y-axes means marker number.
Number of homozygous and heterozygous SNPs and population structure analysis
The number of homozygous and heterozygous SNPs are listed in Additional File A1 and Additional File A2. Plant material (SZ7) had the most homozygous SNPs markers and plant material (SZ110) had the most heterozygous SNPs markers. SZ7 has the highest Ho/He rate. The population structure was calculated using Structure software based on the SNPs data. Parameter K was settled from 3 to 9. The optimal K value was 7 and is shown in Fig. 3. Based on the optimal K value, structural analysis was then conducted using Structure software, and the result was shown in Fig. 4. In all, 108 progenies and 2 parents were clustered into 7 subpopulations.

Optimal K selection. The x-axes means K value and y-axes was mean value of ln likelihood respond to K value.

Population structure. The x-axes indicate individual plant accessions and y-axes means ancestries of each individual plant accessions.
Construction of high-density linkage map
The high-density parental linkage maps were first constructed using Joinmap4.1 software26 (logarithm of odds (LOD) ≥5) to map 1,890 markers for the seed parent and 2,149 markers for the pollen parent. The parental maps contained 17 linkage groups, which were consistent with the haploid chromosome number of hawthorn10.
For the seed parent linkage map, 1,890 markers were mapped to 1,780 distinct positions. The total linkage map length was 1,266.81 cM, the longest linkage group, LG7, was 107.93 cM, the shortest linkage group, LG17, was 34.43 cM, and the average linkage group length was 74.52 cM. LG6, which contained 117 markers, had the highest number of markers, while LG17 contained the lowest number of markers at 15; the average marker number for these 17 linkage groups was 111. The longest average marker interval of 2.46 cM was found in LG17, the shortest average marker interval of 0.38 cM was found in LG6, and the average marker interval of these 17 linkage groups was 0.68 cM. The percentage of ‘Gap ≤5′ was used to reflect the linkage level between adjacent markers in the same linkage group. In the seed parent map, LG15 had the lowest percentage (82.05), while LG14 had the highest percentage (100.00) (Table 3 and Fig. 5).

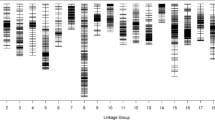
Genetic lengths and marker distribution in 17 linkage groups of consensus linkage map. A black bar means a 2b-RAD marker. The left scale plate imeans genetic distance (centiMorgan as unit).
For the pollen parent linkage map, 2,149 markers were mapped to 2,015 distinct positions. The total linkage map length was 1257.02 cM, the longest linkage group, LG8, was 97.86 cM, the shortest linkage group, LG17, was 14.23 cM, and the average linkage group length was 73.94 cM. LG3, which contained 224 markers, had the highest number of markers, while LG17 contained the lowest number of markers at 12; the average marker number for these 17 linkage groups was 126. The longest average marker interval of 1.29 cM was found in LG17, the shortest average marker interval of 0.28 cM was found in LG2, and the average marker interval of these 17 linkage groups was 0.59 cM. In the pollen parent map, LG17 had a lowest percentage of ‘Gap ≤5′ (90.00), while LG3 had the highest percentage (99.54) (Table 3 and Fig. 6).

Genetic lengths and marker distribution in 17 linkage groups of pollen parent linkage map. A black bar means a 2b-RAD marker. The left scale plate imeans genetic distance (centiMorgan as unit).
The hawthorn consensus linkage map was constructed by integrating the two parental maps based on 823 shared markers. It contained 3,894 markers that were mapped to 3,296 distinct positions. The total linkage map length was 1,551.97 cM, the longest linkage group was LG4 (150.37 cM), the shortest linkage group was LG17 (35.03 cM), and the average linkage group length was 91.29 cM. LG1 contained the highest number of markers at 327, LG17 contained the lowest number of markers at 16, and the average number of markers for these 17 linkage groups was 229. The longest average marker interval of 2.34 cM was found in LG17, the shortest average marker interval of 0.30 cM was found in LG1, and the average marker interval of these 17 linkage groups was 0.40 cM. In the consensus map, LG17 had the lowest percentage of ‘Gap ≤5′ (92.86), while LG5 had the highest percentage (100.00) (Table 4 and Fig. 7).

Genetic lengths and marker distribution in 17 linkage groups of seed parent linkage map. A black bar means a 2b-RAD marker. The left scale plate imeans genetic distance (centiMorgan as unit).
QTL mapping analysis of hawthorn flavonoid content
According to the analysis of five flavonoid component contents in 2014 and 2015, vitexin-rhamnoside was the major component followed by hyperoside, rutin, vitexin and quercetin. The concentrations of vitexin-rhamnoside were 0.00–0.849% and 0.00–0.825% in 2014 and 2015, respectively; the concentrations of hyperoside were 0.00–0.327% and 0.00–0.204% in 2014 and 2015, respectively; the concentrations of rutin were 0.00–0.037% and 0.00–0.078% in 2014 and 2015, respectively; the concentrations of vitexin were 0.00–0.038% and 0.00–0.039% in 2014 and 2015, respectively; and the concentrations of quercetin were 0.00–0.003% and 0.00–0.010% in 2014 and 2015, respectively(Additional File A4).
QTL mapping was conducted for hawthorn leaf flavonoid content, which was measured in 2014 and 2015. In total, 21 QTLs located in 10 linkage groups affected the flavonoid content (Table 5 and Fig. 8).

Genomic QTL distribution on 10 different chromosomes.
qVR15a, which is located in LG8, is related to vitexin-rhamnoside content and accounted for 17.70% of the variance in 2015; qV14a, qV14b, and qV14c are in LG6, LG14, and LG16, respectively; these are related to vitexin content, and in 2014, they explained 16.30–59.00% of the variance. In addition, qV15a, qV15b, qV15c, and qV15d, which are located in LG1, LG6, LG10, and LG16, respectively, are also related to vitexin content; in 2015, they explained 16.70–52.60% of the variance.
qR14a, qR14b, and qR14a, which were located in LG6, LG9, and LG15, respectively, were related to rutin content; in 2014, they explained 17.40–21.40% of the variance. qR15a, which was located in LG7, was also related to rutin content and in 2015 accounted for 25.00% of the variance. The linkage group qH14a, which is located in LG1, is related to hyperoside content and in 2014 accounted for 17.30% of the variance. qH15a, qH15b, and qH15c, which are located in LG3, LG6, and LG7, respectively, are related to hyperoside content, and in 2015, they explained 18.20–21.10% of the variance.
qQ14a, qQ14b, and qQ14c, which are located in LG1, LG3, and LG7, respectively, are related to quercetin content; in 2014, they explained 20.80–21.80% of the variance. Lastly, qQ15a and qQ15b, which are located in LG7 and LG8, respectively, are also related to quercetin content; in 2015, they explained 24.10–43.60% of the variance.
Discussion
Genotyping with molecular markers is useful for studies on phylogeny, evolution, plant breeding, and diseases27,28,29. Restriction fragment length polymorphisms (RFLPs), randomly amplified polymorphic DNA (RAPD), amplified fragment length polymorphisms (AFLPs), and simple sequence repeats (SSRs) were once the mainstays of genotyping. However, because they are based on gel electrophoresis, these methods usually require a substantial amount of time, with high labour costs, to sample a large population. In addition, these methods are prone to error from artificial bands. Moreover, because of marker number limitations, these methods were not suitable for constructing high-density linkage maps, assembling chromosomes, constructing fine-scale QTL maps, and breeding with marker-assisted selection30,31,32,33.
The emergence and development of NGS technology has made RAD-seq a feasible route to genotype the entire genome in a short time. However, library construction for RAD-seq is labour-intensive and time-consuming, and thus modifications such as ddRAD34, SLAF35, 36, and 2b-RAD23 have been introduced. As a high-throughput sequencing technique, 2b-RAD can be used for large-scale genotyping, and compared to traditional molecular marker sequencing techniques, it can construct the linkage map with high marker density and good uniformity. The 2b-RAD method has many advantages. It can provide a streamlined alternative to existing RAD-seq library construction methods, because all reactions occur consecutively in a single well within 4 h. At the same time, 2b-RAD can detect almost every restriction site in the genome in parallel, whereas other RAD-seq methods can only detect a subset of sites. The third advantage that 2b-RAD provides is a choice of selective adaptors, which can adjust to the marker density in the genome. This choice can balance the level of genotyping detail against sequencing throughput capabilities, depending on the type of study37.
In this study, we constructed the refined high-density linkage map for hawthorn using the 2b-RAD method. Other studies have reported the construction of high-density linkage maps using the 2b-RAD method. Guo et al.38 found 1,385 SNP markers in rice, Tian et al.39 found 7,389 SNPs in sea cucumber, and Fu et al.40 found 3,121 SNPs in their high-density linkage map for bighead carp. Compared to linkage maps constructed using RAD and GBS methods16, 17, 41,42,43,44, whose marker numbers ranged from several hundreds to thousands, the linkage map in this study contained 3,894 SNP markers, comparable to and suitable for QTL mapping of flavonoid content. Moreover, these mapped markers can also be used for candidate gene discovery and de novo chromosome assembly for hawthorn.
We first conducted fine-scale QTL mapping analysis of flavonoid content for hawthorn leaves based on this high-density linkage map. Flavonoid content was measured in 2014 and 2015, and 21 QTLs related to flavonoid content were discovered in 10 linkage groups. This preliminary QTL mapping analysis for flavonoid content in hawthorn leaves will continue, especially for the QTLs that were adjacent within a single linkage group. Both qV14c and qV15d were related to vitexin content, had high LOD values (17.4 and 14.59, respectively), and explained 59.00% and 52.60% of the variance, respectively; these QTLs will be the focus of future research.
Materials and Methods
Plant material and DNA extraction
The diploid seed parent material came from cv. Shandongdamianqiu, the most common cultivar in Shandong Province and Beijing, China. The diploid pollen parent came from cv. Damianqiu, which is a cultivar native to Anshan city in northeast China. The parents were hybridized to produce 107 progenies (is it including direct and reciprocal cross progenies?). Genomic DNA was extracted using the CTAB method, and RNA digestion was conducted by adding the proper quantities of RNase and incubating at 37 °C for 30 min. DNA samples were checked for quality and concentration and used for further experiments.
Flavonoid content determination
In 2014 and 2015, the content of five flavonoid monomers, vitexin-rhamnoside, vitexin, rutin, hyperoside, and quercetin, was detected using an Agilent 1100 HPLC with a DAD detector and a C18 column (250 × 4.6 mm, 5 μm). Acetonitrile (A) and 0.5% phosphoric acid solution (B) were used as the mobile phase with the following gradient elution protocol: 0–9 min, 18–20%(A); 9–25 min, 20–50%(A); 25–33 min, 50–18%(A); 33–38 min, 18%(A). The maximum absorption spectrum was 345 nm, the column temperature was 25 °C, the flow rate was 1.0 mL·min−1, and the injection volume was 20 μL.
Library construction and sequencing
The 2b-RAD libraries were prepared for 109 samples by following the protocol developed by Wang et al.23. A total of 100 ng genomic DNA was digested by 4 U BsaXI (New England Biolabs) in a 15-ml reaction at 37 °C for 3 h. A 1% agarose gel was used to verify the digestion of genomic DNA (~30 ng). A total of 12 ml ligation master mix containing 0.2 mM each of two library-specific adaptors, 1 mM ATP (New England Biolabs), and 800 U T4 DNA ligase (New England Biolabs) was added to the digestion product at 4 °C for 16 h, and the mixture was inactivated by heat at 65 °C for 20 min. Ligation products were amplified in three 20 ml reactions per sample, each composed of 7 ml ligated DNA, 0.1 mM each primer, 0.3 mM dNTPs, 1× Phusion HF buffer, and 0.4 U Phusion high-fidelity DNA polymerase (New England Biolabs). PCR was conducted in a DNA Engine Tetrad 2 thermal cycler (Bio-Rad) with 20 cycles of 98 °C for 5 s, 60 °C for 20 s, and 72 °C for 10 s, with a final extension at 72 °C for 10 min. A 2% agarose gel was used for excising the target band, and the DNA will be diffused into nuclease-free water at 4 °C for 12 h. Sample-specific barcodes were introduced by PCR with platform-specific barcode-bearing primers. Each 20 ml PCR reaction contained 25 ng of gel-extracted PCR product, 0.1 mM of each primer, 0.3 mM dNTPs, 1× Phusion HF buffer, and 0.4 U Phusion high-fidelity DNA polymerase; four or five cycles of the PCR profile listed above were performed. PCR products were purified using a QIAquick PCR purification kit (Qiagen) and pooled for sequencing using the Illumina HiseqXTen platform.
Sequence data pre-processing and de novo genotyping
Genotyping was performed using procedures described by Jiao et al.37 Raw reads were first trimmed to remove adaptor sequences. The 3′ terminal positions were excluded from each read to eliminate artefacts that might have arisen at ligation sites. Reads with no restriction sites or containing ambiguous base calls (Ns), long homopolymer regions (>10 bp), and regions with more than five consecutive low-quality (score <10) positions were removed. The remaining trimmed, high-quality reads formed the basis of subsequent analyses. De novo 2b-RAD genotyping was performed using the program RADtyping v1.0.
Linkage map construction
lm × ll (markers from the pollen parent) or nn × np (markers from the seed parent) were categorized as the dominant markers, which segregated in a 1:1 ratio in the map population. Next, hk × hk (markers in both parents) was categorized as co-dominant markers, which were present in both parents and segregated in a ratio of 1:2:1. Before grouping, the markers were tested for goodness-of-fit based on the expected Mendelian ratios through chi-square tests to eliminate markers that significantly deviated from the expected ratios (p-value ≤ 0.05). The qualified markers were then used to construct paternal and maternal linkage maps using the JoinMap 4.1 software26. An LOD score cut-off of 5.0 was used to determine the genetic positions of the markers. Map distances (cM) were converted using recombination frequencies through the Kosambi mapping function. The consensus map was generated by integrating the parental maps based on the shared markers using MergeMap44, 45 with a map weight a 1.0. The visualized linkage maps were subsequently drawn using MapChart 2.246.
QTL mapping for content of five flavonoids
QTL mapping analysis was performed for the content of five flavonoids in hawthorn using MapQTL47. The LOD scores were first analysed using the interval mapping model; LOD statistics were calculated at an interval of 1 cM. Genome-wide and chromosome-wide LOD significance thresholds at the 95% level were determined with a 1000 permutation test for the content of five flavonoids, and QTLs with LOD scores greater than the LOD threshold at 95% were declared significant. Once a QTL was detected, the confidence interval was calculated using the protocol of Li48.
References
Xie, Y. R. et al. The chemical analysis of Shan Li Hong and comparison of the truits of grataegue species in China. Journal of Integrative Plant Biology 23, 383–388 (1981).
Gao, G. Y. & Feng, Y. X. Analysis of the chemical constituents of Hawthorn fruits and their quality evaluation. Acta Pharmaccutica sinica 30, 138–143 (1995).
Nikolov, N., Seligmann, O., Wagner, H. & Horowitz, R. M. Gentili BN. Neue Flavonoid-Glykoside aus Crataegus monogyna und Crataegus pentagyna. Planta Medica 44, 50–53 (1982).
Yang, B. & Liu, P. Composition and health effects of phenolic compounds in hawthorn (Crataegus spp.) of different origins. Journal of the Science of Food & Agriculture 92, 1578–1590 (2012).
Tao, W. Regulation effects of Crataegus pinnatifida leaf on glucose and lipids metabolism. Journal of Agricultural & Food Chemistry 59, 4987–4994 (2011).
Rodrigues, S. et al. Crataegus monogyna buds and fruits phenolic extracts: Growth inhibitory activity on human tumor cell lines and chemical characterization by HPLC–DAD–ESI/MS. Food Research International 49, 516–523 (2012).
Chang, Q., Zuo, Z., Francisco Harrison, M. D. & Chow, M. S. S. Hawthorn. Journal of Clinical Pharmacology 42, 605–612 (2002).
Dixon, R. A. & Paiva, N. L. Stress-Induced Phenylpropanoid Metabolism. Plant Cell 7, 1085–1097 (1995).
Dixon, R. A. & Steele, C. L. Flavonoids and isoflavonoids – a gold mine for metabolic engineering. Trends in Plant Science 4, 394–400 (1999).
Wang, G., Guo, Y., Zhao, Y., Su, K. & Zhang, J. J. Construction of a molecular genetic map for hawthorn based on SRAP markers. Biotechnology & Biotechnological Equipment 29, 1–7 (2015).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics 12, 499–510 (2011).
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. & Johnson, E. A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research 17, 240–248 (2007).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS One 7, e37135 (2012).
Sun, X. et al. SLAF-seq: An Efficient Method of Large-Scale De Novo SNP Discovery and Genotyping Using High-Throughput Sequencing. PLoS One 8, e58700 (2013).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS One 7, 251–264 (2012).
Pfender, W. F., Saha, M. C., Johnson, E. A. & Slabaugh, M. B. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Tagtheoretical & Applied Geneticstheoretische Und Angewandte Genetik 122, 1467–1480 (2011).
Chutimanitsakun, Y. et al. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. Bmc Genomics1 2, 4 (2011).
Wang, N., Fang, L., Xin, H., Wang, L. & Li, S. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. Bmc Plant Biology 12, 1–15 (2007).
Zhang, Z., Wei, T., Zhong, Y., Li, X. & Huang, J. Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genetics & Genomes 12, 1–10 (2016).
Qi, Z. et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS One 9, e104871 (2014).
Zhang, Y. et al. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. Bmc Plant Biology 13, 1–12 (2013).
Liu., T. et al. Construction of the first high-density genetic linkage map ofSalvia miltiorrhizausing specific length amplified fragment (SLAF) sequencing. Scientific Reports 6 (2016).
Wang, S., Meyer, E., Mckay, J. K. & Matz, M. V. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nature Methods 9, 808–810 (2012).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Fu, X. et al. RADtyping: An Integrated Package for Accurate De Novo Codominant and Dominant RAD Genotyping in Mapping Populations. PLoS One 8, 995–998 (2013).
Stam, P. Construction of integrated genetic linkage maps by means of a new computer package: Join Map. Plant Journal 3, 739–744 (1993).
Avise, J. C. Molecular Markers, Natural History and Evolution New York:Chapman and hall; (1994).
Mohan, M. et al. Genome mapping, molecular markers and marker-assisted selection in crop plants. Molecular Breeding 3, 87–103 (1997).
Sidransky, D. Emerging molecular markers of cancer. Nature Reviews Cancer 2, 210–219 (2002).
West, M. A. L. et al. High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Research 16, 787–795 (2006).
Kennedy, G. C. et al. Large-scale genotyping of complex DNA. Nat Biotechnol. Nature Biotechnology 21, 1233–1237 (2003).
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. Genome Research 19, 1068–1076 (2009).
Sun, X., Habier, D., Fernando, R. L., Garrick, D. J. & Dekkers. J. C. Genomic breeding value prediction and QTL mapping of QTLMAS2010 data using Bayesian Methods. Bmc Proceedings.5 Suppl 3(Suppl 2):1-8 (2011).
Kai, W. et al. A ddRAD-based genetic map and its integration with the genome assembly of Japanese eel (Anguilla japonica) provides insights into genome evolution after the teleost-specific genome duplication. Bmc Genomics 15, 233 (2014).
Wei, Q. et al. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. Bmc Genomics 15, 1–10 (2014).
Guo, Y. S. et al. Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L. × Vitis amurensisRupr.). Frontiers in Plant Science 6, 393 (2015).
Jiao, W. et al. Editor’s choice: High-Resolution Linkage and Quantitative Trait Locus Mapping Aided by Genome Survey Sequencing: Building Up An Integrative Genomic Framework for a Bivalve Mollusc. Dna Research 21, 85–101 (2013).
Guo, Y. et al. An improved 2b-RAD approach (I2b-RAD) offering genotyping tested by a rice (Oryza sativa L.) F2 population. Bmc Genomics 15, 956 (2014).
Tian, M. et al. Construction of a High-Density Genetic Map and Quantitative Trait Locus Mapping in the Sea Cucumber Apostichopus japonicus. Scientific Reports 5 (2015).
Fu, B., Liu, H., Yu, X. & Tong, J. A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Scientific Reports 6 (2016).
Ward, J. A. et al. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. Bmc Genomics 14, 2 (2013).
Gonen, S. et al. Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. Bmc Genomics 15, 166 (2014).
Yang, H. et al. Application of next-generation sequencing for rapid marker development in molecular plant breeding: a case study on anthracnose disease resistance in Lupinus angustifolius L. Bmc Genomics 13, 1–12 (2012).
Kakioka, R., Kokita, T., Kumada, H., Watanabe, K. & Okuda, N. A. RAD-based linkage map and comparative genomics in the gudgeons (genus Gnathopogon, Cyprinidae). Bmc Genomics 14, 1–11 (2013).
Wu, Y., Close, T. J. & Lonardi, S. On the accurate construction of consensus genetic maps. Comput Syst Bioinformatics Conf 7, 285–296 (2008).
Voorrips, R. E. MapChart: software for the graphical presentation of linkage maps and QTLs. Journal of Heredity 93, 77–78 (2002).
Ooijen, J. W. V. MapQTL 5. Software for the Mapping of Quatitative Trait Loci in Experimental Population (2004).
Li, H. A quick method to calculate QTL confidence interval. Journal of Genetics 90, 355–360 (2011).
Acknowledgements
Research supported by the National Natural Science Funds of China (grant #31101515) and Natural Science Founds of Liaoning Province of China (grant #20170540804).
Author information
Authors and Affiliations
Contributions
Yuhui Zhao, Jijun Zhang and Yinshan Guo conducted the major part of the research including preparation of the 2bRAD-seq data, bioinformatics analysis and manuscript preparation. Kai Su, Gang Wang, Liping Zhang and Junpeng Li were involved in one or more processes of DNA extraction or bioinformatics analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Su, K., Wang, G. et al. High-Density Genetic Linkage Map Construction and Quantitative Trait Locus Mapping for Hawthorn (Crataegus pinnatifida Bunge). Sci Rep 7, 5492 (2017). https://doi.org/10.1038/s41598-017-05756-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05756-5
- Springer Nature Limited