

Abstract
The linear ubiquitin assembly complex (LUBAC) consists of HOIP, HOIL-1 and SHARPIN and is essential for proper immune responses. Individuals with HOIP and HOIL-1 deficiencies present with severe immunodeficiency, autoinflammation and glycogen storage disease. In mice, the loss of Sharpin leads to severe dermatitis due to excessive keratinocyte cell death. Here, we report two individuals with SHARPIN deficiency who manifest autoinflammatory symptoms but unexpectedly no dermatological problems. Fibroblasts and B cells from these individuals showed attenuated canonical NF-κB responses and a propensity for cell death mediated by TNF superfamily members. Both SHARPIN-deficient and HOIP-deficient individuals showed a substantial reduction of secondary lymphoid germinal center B cell development. Treatment of one SHARPIN-deficient individual with anti-TNF therapies led to complete clinical and transcriptomic resolution of autoinflammation. These findings underscore the critical function of the LUBAC as a gatekeeper for cell death-mediated immune dysregulation in humans.







Similar content being viewed by others

Data availability
RNA sequence data have been deposited in the Gene Expression Omnibus under the accession code GSE261031. Exome sequencing data will not be made publicly available as they contain information that might compromise research participant privacy/consent. Source data are provided with this paper.
References
Iwai, K., Fujita, H. & Sasaki, Y. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat. Rev. Mol. Cell Biol. 15, 503–508 (2014).
Fuseya, Y. et al. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 22, 663–673 (2020).
Kelsall, I. R., Zhang, J., Knebel, A., Arthur, J. S. C. & Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl Acad. Sci. USA 116, 13293–13298 (2019).
Boisson, B. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939–951 (2015).
Boisson, B. et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13, 1178–1186 (2012).
Oda, H. et al. Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front. Immunol. 10, 479 (2019).
Peltzer, N. et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117 (2018).
Peltzer, N. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165 (2014).
HogenEsch, H. et al. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 143, 972–982 (1993).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471, 637–641 (2011).
Kumari, S. et al. SHARPIN prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 3, e03422 (2014).
Rickard, J. A. et al. TNFR1-dependent cell death drives inflammation in SHARPIN-deficient mice. eLife 3, e03464 (2014).
Tokunaga, F. et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).
Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Sasaki, Y. et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. EMBO J. 32, 2463–2476 (2013).
Draber, P. et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 13, 2258–2272 (2015).
Vince, J. E. et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 (2007).
de Almagro, M. C., Goncharov, T., Newton, K. & Vucic, D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 6, e1800 (2015).
Laurien, L. et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11, 1747 (2020).
Kay, J. & Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology 43, iii2–iii9 (2004).
Wijbrandts, C. A. et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann. Rheum. Dis. 67, 1139–1144 (2008).
Sundberg, J. P. et al. Keratinocyte-specific deletion of SHARPIN induces atopic dermatitis-like inflammation in mice. PLoS ONE 15, e0235295 (2020).
Wang, J. et al. LUBAC suppresses IL-21-induced apoptosis in CD40-activated murine B cells and promotes germinal center B cell survival and the T-dependent antibody response. Front. Immunol. 12, 658048 (2021).
Roco, J. A. et al. Class-switch recombination occurs infrequently in germinal centers. Immunity 51, 337–350 (2019).
Elsner, R. A. & Shlomchik, M. J. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity 53, 1136–1150 (2020).
Teh, C. E. et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat. Commun. 7, 13353 (2016).
Park, Y. et al. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat. Immunol. 17, 286–296 (2016).
HogenEsch, H. et al. Increased expression of type 2 cytokines in chronic proliferative dermatitis (cpdm) mutant mice and resolution of inflammation following treatment with IL-12. Eur. J. Immunol. 31, 734–742 (2001).
van Zelm, M. C. et al. Human CD19 and CD40L deficiencies impair antibody selection and differentially affect somatic hypermutation. J. Allergy Clin. Immunol. 134, 135–144 (2014).
Meyers, G. et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc. Natl Acad. Sci. USA 108, 11554–11559 (2011).
McGowan, H. W. et al. Sharpin is a key regulator of skeletal homeostasis in a TNF-dependent manner. J. Musculoskelet. Neuronal Interact. 14, 454–463 (2014).
Kim, H. et al. Development of a validated interferon score using NanoString technology. J. Interferon Cytokine Res. 38, 171–185 (2018).
Panayotova-Dimitrova, D. et al. cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 5, 397–408 (2013).
Weinlich, R. et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 5, 340–348 (2013).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Sarhan, J. et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA 115, E10888–E10897 (2018).
Gurung, P., Lamkanfi, M. & Kanneganti, T. D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 194, 2064–2067 (2015).
Douglas, T., Champagne, C., Morizot, A., Lapointe, J. M. & Saleh, M. The inflammatory caspases-1 and -11 mediate the pathogenesis of dermatitis in SHARPIN-deficient mice. J. Immunol. 195, 2365–2373 (2015).
Gurung, P., Sharma, B. R. & Kanneganti, T. D. Distinct role of IL-1β in instigating disease in Sharpin(cpdm) mice. Sci. Rep. 6, 36634 (2016).
Anderton, H. et al. Langerhans cells are an essential cellular intermediary in chronic dermatitis. Cell Rep. 39, 110922 (2022).
Anderton, H., Wicks, I. P. & Silke, J. Cell death in chronic inflammation: breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 16, 496–513 (2020).
van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat. Rev. Immunol. 23, 289–303 (2023).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571 (2020).
Weisel, K. et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 23, 85 (2021).
Weisel, K. et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 8, e000680 (2021).
Lalaoui, N. et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108 (2020).
Tao, P. et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114 (2020).
Cuchet-Lourenco, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 116, 970–975 (2019).
Taft, J. et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell 184, 4447–4463 (2021).
Badran, Y. R. et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J. Exp. Med. 214, 1937–1947 (2017).
Damgaard, R. B. et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11, e9324 (2019).
Zinngrebe, J. et al. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 213, 2671–2689 (2016).
Kelsall, I. R. et al. HOIL-1 ubiquitin ligase activity targets unbranched glucosaccharides and is required to prevent polyglucosan accumulation. EMBO J. 41, e109700 (2022).
Otten, E. G. et al. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 594, 111–116 (2021).
Matsumoto, M. L. et al. Engineering and structural characterization of a linear polyubiquitin-specific antibody. J. Mol. Biol. 418, 134–144 (2012).
Zinngrebe, J. et al. Compound heterozygous variants in OTULIN are associated with fulminant atypical late-onset ORAS. EMBO Mol. Med. 14, e14901 (2022).
Samson, A. L. et al. A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells. Cell Death Differ. 28, 2126–2144 (2021).
Wang, K. et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell 180, 941–955 (2020).
Acknowledgements
We thank the participants and their family members and the healthy donors for their enthusiastic support during this research study and M. Pasparakis, H. Kashkar and A. Annibaldi (Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases and Center for Molecular Medicine Cologne, University of Cologne, Germany) for logistic support and valuable suggestions. We thank the The Walter and Eliza Hall Institute Histology Core Facility for their support and assistance in this work. This study was supported, in part, by the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institute of Allergy and Infectious Diseases, the National Cancer Institute, the National Heart, Lung, and Blood Institute, the Clinical Center of the NIH and the Peter MacCallum Cancer Foundation. H.O. was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation), including CRC1403 (414786233) and Germany’s Excellence Strategy (EXC 2030 (390661388)) and by Fritz-Thyssen Stiftung (10.23.1.013MN). K.E.L. is supported by an Australian Research Council Future Fellowship (FT190100266). N.L., J. Silke and H.A. are supported by National Health and Medical Research Council Leadership Investigator Grants (2017929, 1195038 and 1194144). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council IRIISS (GNT9000719). S.B. is supported by France’s National Research Agency, the Investment for the Future Program (ANR-11-LABX-0070_TRANSPLANTEX) as well as Strasbourg’s Interdisciplinary Thematic Institute for Precision Medicine, CNRS and INSERM, funded by IdEx Unistra (ANR-10-IDEX-0002) and SFRI-STRAT’US (ANR-20-SFRI-0012). H.W. is supported by Cancer Research UK (A27323), a Wellcome Trust Investigator Award (214342/Z/18/Z), the Medical Research Council (MR/S00811X/1), the DFG (CRC1399, 413326622; CRC1530, 455784452; CRC1403, 414786233), an Alexander von Humboldt Foundation Professorship Award and CANTAR. The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, the NIH (R21AI159728 to B.B. & P01AI061093 to J.L.C.), the National Center for Advancing Translational Sciences (NCATS), NIH Clinical and Translational Science Award (CTSA) program (UL1TR001866), the French National Research Agency (ANR) under the ‘Investments for the Future’ program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French Foundation for Medical Research (FRM) (EQU201903007798), the Square Foundation, Grandir – Fonds de solidarité pour l’enfance, William E. Ford, General Atlantic’s Chairman and Chief Executive Officer, Gabriel Caillaux, General Atlantic’s Co-President, Managing Director and Head of Business in EMEA, and the General Atlantic Foundation, Institut National de la Santé et de la Recherche Médicale (INSERM) and the University of Paris Cité.
Author information
Authors and Affiliations
Contributions
H.O., N.L., I.A. and D.L.K. conceived and designed the study, analyzed the data and wrote the manuscript. K.M., P.P.C., E.R., O.V., O.K., C.R., S.N., H.S.K., M. Swart, Y.W., N.I.Ç., A.M., R.C., Q.X., S.P., D.B.B., J.J.C., K.D., C.L.S., H.A., K.E.L., H.Y., D.Y., M.B., D.R., W.L.T., M.G. and J.T. performed experiments. B.M., J.M., J. Stoddard and J.N. analyzed and interpreted the results. K.M., M.N., A.K.O., P.H., T.R., N.T.D., H.K., V.Z., N.M., M. Shahrooei, N. Parvaneh, N.A.-O., R.C., J.D., P.M., M.J.K., B.B., J.-L.C., S.B. and A.P.R. developed the human materials and/or recruited participants. C.T.M., K.I., S.D.R., L.D.N., J. Silke, P.L.S., N. Peltzer and H.W. provided critical scientific input and/or reagents. H.O. and N.L. wrote the initial draft of the paper. All authors contributed to the final review and editing of the paper.
Corresponding authors
Ethics declarations
Competing interests
S.P. is currently an employee of AstraZeneca and may own stock or stock options. The other authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks Geert van Loo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: N. Bernard, in collaboration with the Nature Immunology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
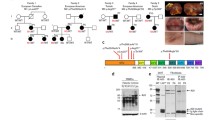
Extended Data Fig. 1 Genetic and molecular investigations of sharpenia.
(a) Sanger sequence electropherograms demonstrating homozygous frameshift variants in the patients. (b) Population allele frequency and CADD score for SHARPIN variants homozygous in public databases. The two SHARPIN variants appear in red. CADD−Mutation Significance Score (MSC) cutoff for SHARPIN (90% confidence interval) was indicated by dashed line. NR: not reported. (c, d) Normalized mRNA levels of SHARPIN in (c) PBMCs and (d) fibroblasts from LUBAC-deficient patients and healthy controls. RNA was extracted from each sample and was measured with technical quadruplicates. Mean value is displayed as a bar. (e, f) Supporting data for Fig. 2e. TNFR1-signaling complex (TNFR1-SC) formation in fibroblasts from P1 and two unrelated healthy controls. Fibroblasts were stimulated with modified tandem affinity purification (moTAP)-tagged TNF (1 μg/ml) for the indicated times. TNFR1-SC was purified with anti-FLAG immunoprecipitation, and analyzed by western blotting. (g) Normal induction of non-canonical NF-κB in P1. Total PBMCs were stimulated with anti-CD3 (aCD3) for the indicated durations, and the expression of NFKB2 p100 (full length) and p52 (active form) was detected by western blot. Sis: P1’s sister carrying a heterozygous p.Leu74ProfsX86 variant. Representative result of two independent experiments.
Extended Data Fig. 2 Cytokine expression studies ex vivo in LUBAC-deficient patients.
(a) Cytokine expression in LUBAC-deficient monocytes. PBMCs from SHARPIN (P1)- and HOIP-deficient patients and two healthy controls were stimulated with IL-1β (10 ng/ml) for 6 h, and the intracellular accumulation of cytokines in CD14+ monocytes was quantified by flow cytometry. (b, c) Cytokine secretion from LUBAC-deficient PBMCs. PBMCs from SHARPIN and HOIP-deficient patients and two healthy controls were stimulated with (b) LPS (1 μg/ml) or (c) IL-1β (10 ng/ml) for 6 h, and secreted cytokines were measured by ELISA. (d, e) Cytokine secretion from LUBAC-deficient fibroblasts. Fibroblasts from SHARPIN (P1)- and HOIP-deficient patients and two healthy controls were stimulated with (d) LPS (1 μg/ml) or (e) IL-1β (10 ng/ml) for 24 h, and secreted cytokines were measured by ELISA. (a-e) The experiments were performed with biological triplicates (a,d,e) or duplicates (b,c), and shown are the representatives of two independent experiments. Mean values ± s.d are displayed.
Extended Data Fig. 3 Cell death induction assays.
(a) Cell death assay using immortalized mouse embryonic fibroblasts from Sharpin-deficient mice stably reconstituted with wild-type SHARPIN or patient-derived SHARPIN mutants (P1 and P2). (b, c) Cell death assay using fibroblasts from a SHARPIN-deficient patient (P1), patients with HOIL1 deficiency, otulipenia and cleavage resistant RIPK1-induced autoinflammation (CRIA), and two unrelated healthy controls. The cells were stimulated with TNF (100 ng/ml) combined with (b) smac mimetic (SM: compound A: 100 nM) or (c) human recombinant TWEAK (50 ng/ml), in the presence or absence of zVAD (pan-caspase inhibitor: 20 μM) or Nec1 (RIPK1 inhibitor: 50 μM). The dead cell percentages after 16 h of treatment are shown. (a-c) The experiments were performed with biological triplicates (a, b) or duplicates (c), and shown are the representatives of two (a, c) or five (b) independent experiments. Mean values ± s.d are displayed. Quantitative data were analyzed using one-way ANOVA followed by Tukey-Kramer test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, N.S., not significant. (d) Supporting data for Fig. 3c. Western blot analysis of caspase-3 cleavage in cell lysates from fibroblasts from a SHARPIN-deficient patient and two unrelated healthy controls. The cells were stimulated with TNF (100 ng/ml) and cycloheximide (CHX 50 μg/ml) for the indicated times. (e, f) Necroptosis induction assay using EBV-immortalized lymphoblastoid cells (e) and fibroblasts (f) from SHARPIN-deficient patient. The fibroblasts were stimulated with TSZ (TNF, smac mimetic (compound A) and zVAD) for the indicated time. HT29 cells were used as a positive control for the phospho-antibodies. Representative result of two independent experiments. (g) Supporting data for Fig. 3d. Complex II immunoprecipitation in fibroblasts from SHARPIN-deficienct P1 compared with an unrelated healthy control. Fibroblasts were stimulated with TNF + zVAD for the indicated times, and the lysates were subjected to immunoprecipitation.
Extended Data Fig. 4 In vivo and in vitro characterization of cell death in human LUBAC deficiency.
(a) Hematoxylin and eosin (H&E) staining of colon biopsy samples from LUBAC-deficient patients. Bars: 0.4 mm. These images are representative of three biopsy specimens per donor. (b, c) Supporting data for Fig. 3g to validate the specificity of p-RIPK1 antibody to detect RIPK1 Ser166 phosphorylation with (b) western blot and (c) immunocytochemistry. HT29 cells were stimulated with TSZ (TNF + smac mimetic (BV6) + zVAD) for 4 h. Note that TSZ-stimulated cells show positive staining of pRIPK1, which was removed by λ-phosphatase treatment. (d, e) Supporting data for Fig. 3h to validate the antibody specificity for cleaved GSDMD (Asp275) with (d) western blot and (e) immunocytochemistry. THP1 cells were pre-incubated with LPS for 3 h and were further stimulated with nigericin for another 1 h. (b-e) These experiments were aimed to confirm the specificity of the antibodies and were not repeated. (f) Cleavage of GSDME in dermal fibroblasts stimulated with TNF (100 ng/ml) + CHX (50 μg/ml) for the indicated times. HeLa cells were used as a positive control. Representative result of two independent experiments.
Extended Data Fig. 5 Characterization of joint inflammation in SHARPIN deficiency.
(a) Multiplex ELISA measurement of chemokines in the sterile synovial fluid from P1 before the initiation of anti-TNF treatment, compared with osteoarthritis (OA) control donors (N = 7). The samples were measured in technical triplicate (P1) or duplicate (OA), respectively. Mean values ± s.d are displayed. (b) Quantification of CD45 positive cells in tendons of shoulder joints of control and Sharpin-deficient mice (n = 4 for each group). Data are represented as mean values + SEM. Significance calculated with a two-tailed Mann-Whitney test. (c) Representative hematoxylin and eosin (H&E) staining sections of elbow joints from Sharpin-deficient mice (N = 2) and wild-type littermate controls (N = 2). The arrowhead indicates an inflamed ligament.
Extended Data Fig. 6 Characterization of secondary lymphoid organs in LUBAC deficiencies.
(a, b) Aberrant formation of lymphoid follicles and paracortex in secondary lymphoid organs from LUBAC deficient patients. (a) Lymph node histology of a HOIP-deficient patient compared with a control specimen from an unrelated donor. (b) Adenoid histology of SHARPIN-deficient P1 compared with a control specimen from an unrelated donor. The immunohistochemistry staining was not repeated due to the limited clinical specimens. (c) Gating strategy for the adenoid spectral flow cytometry analysis.
Extended Data Fig. 7 Spectral flow cytometry analysis of human adenoids.
(a–e) Adenoid single-cell suspensions from SHARPIN-deficient P1 and 10 unrelated pediatric control donors were analyzed. (a) Quantification of CD3+, CD4+, CD8+ and CD20+ populations in adenoid samples. (b, c) Surface immunoglobulin expression in the (b) germinal center B and (c) memory B populations. (d, e) Quantification of T cell subpopulations in the adenoid samples. Mean values ± s.d are displayed.
Extended Data Fig. 8 Normal T cell phenotyping results in the SHARPIN-deficient patient ex vivo.
(a) T cell proliferation assay. PMBCs were incubated with Cell Trace Violet, stimulated with anti-CD3/28 or PHA for 72 h and analyzed by flow cytometer. (b) Intracellular cytokine staining for Th1, Th2 and Th17 populations. PBMCs were stimulated with PMA (100 ng/ml) and ionomycin (1 μM) for 5 h with Brefeldin A. Stimulated cells were surface stained, fixed and permeabilized with BD Cytofix/Cytoperm kit. Cells were further stained for intracellular cytokines and analyzed by flow cytometry. Ctrl: unrelated healthy control, Sister: sister carrying the heterozygous frameshift SHARPIN variant p.Leu74ProfsX86. Representative result of two independent experiments.
Extended Data Fig. 9 Whole blood RNA sequencing.
(a, b) mRNA expression of selected cytokines (a) and chemokines (b) in the pre- and post-anti-TNF treatment P1 whole blood RNA samples as well as four age-matched healthy controls. (c) A heatmap demonstrating the changes of genes representative for type I interferon-stimulated gene signature in pre- and post-treatment samples from the SHARPIN-deficient P1.
Supplementary information
Source data
Source Data
Unprocessed western blots.
Rights and permissions
About this article

Cite this article
Oda, H., Manthiram, K., Chavan, P.P. et al. Biallelic human SHARPIN loss of function induces autoinflammation and immunodeficiency. Nat Immunol 25, 764–777 (2024). https://doi.org/10.1038/s41590-024-01817-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-024-01817-w
- Springer Nature America, Inc.