

Abstract
Emerging evidence implicates common genetic variation - aggregated into polygenic scores (PGS) - in the onset and phenotypic presentation of rare diseases. Here, we comprehensively map individual polygenic liability for 1102 open-source PGS in a cohort of 3059 probands enrolled in the Genomic Answers for Kids (GA4K) rare disease study, revealing widespread associations between rare disease phenotypes and PGSs for common complex diseases and traits, blood protein levels, and brain and other organ morphological measurements. Using this resource, we demonstrate increased polygenic liability in probands with an inherited candidate disease variant (VUS) compared to unaffected carrier parents. Further, we show an enrichment for large-effect rare variants in putative core PGS genes for associated complex traits. Overall, our study supports and expands on previous findings of complex trait associations in rare diseases, implicates polygenic liability as a potential mechanism underlying variable penetrance of candidate causal variants, and provides a framework for identifying novel candidate rare disease genes.
Similar content being viewed by others

Introduction
The Genomic Answers for Kids (GA4K) study at Children’s Mercy Research Institute is a large-scale, phenotypically diverse pediatric rare disease cohort comprising patient cases referred from 22 different hospital specialties1,2. Comprehensive clinical genome assessment utilizing structured rare disease phenotypes and prioritized variants from whole-exome or whole-genome sequencing are further combined with additional omic approaches with the goal of improving understanding of the genetics of rare diseases.
One such strategy is the integration of polygenic scores (PGS) to assess the contribution of common genetic variants to rare disease phenotypes. PGS approaches have been previously applied to estimate disease risk attributed to inherited common variant polygenic background—such as in severe neurodevelopmental disorders3; and in integrating trait-matched PGS to understand differences in disease penetrance among carriers of monogenic risk variants4,5,6,7. Further, differences in individual PGS liability can help resolve variable expressivity of complex, multi-phenotype rare disorders, such as risk for schizophrenia among carriers of the 22q11.2 deletion8.
PGS effect estimates have also highlighted a sharing of underlying causal genes in monogenic and matched common diseases and other correlated complex traits9,10, providing opportunities for expanded discovery of large-effect variants underlying rare disease patient cases. Integrating common variant PGSs derived from population-scale resources available from PGS Catalog11, here we systematically mapped the impacts of >1000 PGSs for common complex diseases, laboratory tests, organ morphological, and anthropometric traits in > 500 groupings of probands defined by their rare disease phenotypes.
Results
Systematic assessment of PGS associations across diverse rare disease phenotypes
To map associations between rare disease phenotypes and common complex diseases, traits, and measurements we first generated a filtered set of PGS obtained from PGS Catalog (N PGS = 1102; Methods). Pairwise correlation across the filtered PGS set was low (median absolute correlation coefficient = 0.019 (SD = 0.068)). Individual scores for each PGS were calculated using imputed genotype data in a subset of 3059 probands enrolled in GA4K (Fig. S1; Table 1; Methods). We further defined a set of probands with EUR ancestry (N = 2641) for HPO-PGS association analysis to reflect the demographic background of the majority of individuals currently comprising PGS training cohorts (Methods; Fig. S2). The proportion of PGS variants recovered in the GA4K imputed genotype callset was high (median = 96%; Fig. S3).
We quantified the contribution of each of the 1102 PGS in 626 rare disease phenotype (HPO) case/controls cohorts (N pairwise comparisons = 689,852; HPO median case N = 11; median HPO terms per proband = 6; Fig. S4) using a phenome-wide association study (PheWAS) approach, mapping each PGS to binary HPO phenotype cohorts. For each of these cohorts, we constructed a logistic regression model comprising PGS, sex, and first five principal components of ancestry, and further compared observed results with null distributions from permutation testing (N permutations = 10,000), yielding an empirical P-value for each HPO-PGS pair (Methods). From this approach we identified 897 significant HPO-PGS pairs (FDR 20%) comprising 525 PGS and 154 HPO cohorts.
Categorizing HPO and PGS into tissue- and/or physiology-specific measurements and disorders, we observed the greatest number of associations for nervous system morphology HPOs with brain volume PGSs (N = 95 HPO-PGS pairs) and growth disorder HPOs associated with body measurement PGSs (N = 49 HPO-PGS pairs) (Fig. 1; Supplementary Data 1). Overall, we observed at least one significant (FDR < 20%) PGS association for 1775 of 2641 (67%) probands in the study cohort. Median regression model fit (Nagelkerke’s pseudo-R2) was 0.62% (range = 0.21–3.30%) (Figure S5). At more stringent FDR thresholds, we observed 391 (FDR 10%) and 178 (FDR 5%) significant HPO-PGS pairs. Summary statistics for all tested HPO-PGS pairs are provided in Supplementary Data 2.

PGS terms (x-axis) are categorized using ontology metadata available from PGS Catalog and further colored by measurement (red), disease (green), other trait (blue), biological process (cyan), or sex-specific PGS (orange). Color gradient indicates mean false discovery rate (FDR) within each HPO/PGS category pair. White indicates that no significant associations were observed for the indicated HPO-PGS pair.
We performed a trans-ancestry analysis of available non-EUR GA4K probands (N = 418), observing high concordance of PGS liability across diverse ancestry groups (Fig. S6). We further performed a replication analysis using case data from an external rare disease cohort (Deciphering Developmental Disorders (DDD)) (N = 1416; Methods; Fig. S7). We observed a replication rate for HPO-PGS pairs (defined as matching PGS coefficient direction of effect and nominal P-value (P ≤ 0.05)) of 17% (FDR 20%), 26% (FDR 10%), and 44% (FDR 5%), which increased for larger DDD sample sizes (Fig. S8).
Impact of PGS liability on clinical diagnosis
We next assessed the impact of PGS on clinical diagnostic status for probands with at least one PGS association and who had completed clinical diagnostic testing at time of study (N = 984). We used a logistic regression model to quantify the contribution of an increasing burden of rare disease phenotypes (HPOs) linked to PGS to the likelihood of having a clinical diagnostic status including “diagnostic”, “VUS/GUS”, “negative”, or “other” (partial genotype or partial diagnosis) classification. We repeated this analysis for increasingly stringent proband PGS Z-score thresholds. For each additional rare disease phenotype (HPO) significantly associated with a PGS (FDR 20%), we observed a modest decrease in the likelihood of diagnostic status (Fisher’s Exact Test (two-sided); odds ratio = 0.97 [CI 0.94–0.99], P = 0.01) and increase in the likelihood of VUS/GUS status (Fisher’s Exact Test (two-sided); odds ratio = 1.03 [CI 1.00–1.06], P = 0.009) (Fig. 2). However, for the most stringent PGS Z-score threshold, this effect was increased (Fisher’s Exact Test (two-sided); diagnostic: odds ratio = 0.90 [CI 0.85–0.96], P = 0.001; VUS/GUS: odds ratio = 1.08 [CI 1.01–1.14], P = 0.01). No differences were observed for negative cases across any PGS Z-score threshold. We next asked whether observed results could be explained by potential challenges with clinical diagnosis of more complex patient cases (that is, a relatively larger number of rare disease phenotypes irrespective of any PGS associations). Again, using a logistic regression model, we quantified the likelihood of each clinical diagnostic status associated with a count of all HPOs per proband and observed no significant associations.

Results are displayed across more stringent PGS Z-score thresholds (from bottom to top of each diagnostic category). Dots indicate the odds ratio from logistic regression (two-sided), error bars indicate 95% confidence interval. P-values passing a threshold of P ≤ 0.05 are indicated in figure. No adjustments were made for multiple comparisons.
Increased polygenic liability in VUS probands compared to unaffected carrier parents
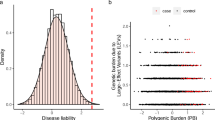
Given previous reports showing modification of monogenic disease penetrance and severity associated with trait-relevant polygenic liability4,6, we hypothesized that significantly associated PGS in probands with a candidate—but presently non-diagnostic—inherited variant in a known rare disease gene (variant of unknown significance (VUS)) would exhibit increased polygenic liability compared with the PGS of their unaffected carrier parent (Fig. 3A). We identified a subset of probands who had completed clinical diagnostic testing which had identified an inherited, autosomal dominant (partial penetrance) VUS and who had significantly associated PGS available for proband, mother, and father (N = 62 HPO-PGS pairs; Methods). We observed a significant difference in associated PGS (FDR 20%) liability between probands and carrier parents (proband median PGS = 0.23, carrier parent median PGS = −0.50; Wilcoxon Rank Sum test (two-sided), P = 5 × 10−04) but not between probands and non-carrier parents (proband median PGS = 0.23, non-carrier parent median PGS = 0.02; Wilcoxon Rank Sum test (two-sided), P = 0.2) (Fig. 3B), where values > 0 indicate increased PGS liability for the associated trait. This effect was more pronounced for more stringent HPO-PGS significance (FDR) thresholds (Fig. 3B). We repeated this analysis for unaffected siblings, where available, observing similar increased PGS liability for probands compared to unaffected carrier siblings (proband median PGS = 0.10, carrier sibling PGS = −0.82; Wilcoxon Rank Sum test (two-sided), P = 8 × 10−05) but not when comparing probands with unaffected non-carrier siblings (proband median PGS = 0.10, non-carrier sibling PGS = 0.13; Wilcoxon Rank Sum test (two-sided), P = 0.94) (Fig. S9A).

a Proposed model of variable rare disease penetrance for inherited candidate pathogenic variant (VUS) in proband and unaffected carrier parent as a function of polygenic liability for an associated complex trait PGS. b Distribution of PGS for probands, carrier (Het) parents, and non-carrier (Ref) parents. PGS with association direction of effect less than zero are inverted to enable visualization. Results are stratified across HPO-PGS significance (FDR) thresholds (from right to left: FDR 20%, 10%, 5%). Cross bars indicate median, error bars indicate standard deviation. P-values are from Wilcoxon Rank Sum test (two-sided). No adjustments were made for multiple comparisons. Results are from 62 HPO:PGS associations across 6 trios (i.e. proband, mother, father) for FDR threshold 20%, 33 HPO:PGS associations across 5 trios for FDR threshold 10%, and 26 HPO:PGS associations across 4 trios for FDR threshold 5%. c Proband minus parent PGS Z-score/standard deviation for significantly associated PGS in probands with a clinical variant of unknown significance (VUS) compared to carrier (Het) and non-carrier (Ref) parents. Points above zero indicate PGS liability is greater in probands and points below zero indicate PGS liability is greater in indicated parent. Pairwise differences for PGS with association direction of effect less than zero are inverted to enable visualization. Diamonds denote median PGS for PGS linked with VUS HPO(s) for probands compared to carrier parents (orange) and probands compared to non-carrier parents (blue). Box and violin plots denote distribution of median PGS for randomly selected PGS not associated with VUS HPO(s), where – for boxplots - the middle line corresponds to the median, the lower and upper edges of the box correspond to the first and third quartiles, the whiskers represent the interquartile range (IQR) ×1.5 and beyond the whiskers are outlier points. P-values are derived from the empirical distribution of these background PGS (N permutations = 10,000). No adjustments were made for multiple comparisons. Results are stratified across HPO-PGS significance (FDR) thresholds (from right to left: FDR 20%, 10%, 5%). Observed results (indicated by diamonds) are from 62 HPO:PGS associations across 6 trios (i.e. proband, mother, father) for FDR threshold 20%, 33 HPO:PGS associations across 5 trios for FDR threshold 10%, and 26 HPO:PGS associations across 4 trios for FDR threshold 5%.
We next performed a within-trio analysis, quantifying the difference in PGS liability between a proband and each of their parents and compared this quantity with repeated random sampling from nominally significant PGS with matching direction of effect and not linked to proband VUS HPOs (N permutations = 10,000; median PGS per proband = 168). PGS liability for PGSs associated with a VUS HPO(s) (FDR 20%) was significantly increased between probands and carrier parents compared to background selected PGS (median difference in PGS liability = 0.58, empirical P = 1 × 10−04), but not between probands and non-carrier parents (median difference in PGS liability = 0.19, empirical P = 0.24) (Fig. 3C). This difference was stronger for more stringent HPO-PGS significance (FDR) thresholds. We repeated this analysis for unaffected siblings, where available, and observed the same trend in PGS liability by carrier/non-carrier status (Fig. S9B).
Enhanced rare disease phenotyping through PGS integration
Next, we used clinical observation data from the electronic medical record (EHR) at the GA4K study site to compare individual PGS with growth trajectories recorded at clinical visits. Previous research has demonstrated that individual polygenic liability for relevant complex traits can be useful for risk stratification of rare disease patient cases12. We observed moderate positive correlation comparing CDC age- and sex-adjusted height and body mass index (BMI) Z-scores (Methods) with a significantly associated PGS for height (PGS ID = PGS000998) (N = 2091; Pearson’s correlation (two-sided), r = 0.36, P < 1 × 10−16) (Fig. 4A) and body mass index (PGS ID = PGS000027) (N = 2091; Pearson’s correlation (two-sided), r = 0.23, P < 1 × 10−16) (Fig. 4B).

a Individual height observations indicate age- and sex-adjusted Z-scores (CDC benchmarks) and are displayed on the y-axis. Individual scores for a significantly associated height PGS are displayed on the x-axis. Red horizontal line indicates short stature threshold (CDC). Blue horizontal line indicates tall stature threshold (CDC). Probands with a short stature HPO in GA4K are indicated by a red triangle if observed height value passes CDC threshold for short stature, or a red cross if not passing this threshold. Probands with tall stature HPO are indicated by a blue triangle if observed height value passes CDC threshold for tall stature, or a blue cross if not passing this threshold. Gray indicates control (no short or tall stature HPO terms present in GA4K). Pearson correlation coefficient test (two-sided) and associated P-value is indicated in figure. Pearson correlation coefficient P = 2 × 10−64. b As in (a) for obesity probands compared with controls, and PGS and growth chart Z-scores for body mass index. Pearson correlation coefficient P = 2 × 10−26.
For height, we further integrated rare disease phenotypes (HPO terms) to annotate “short stature” (HP:0004322), “tall stature” (HP:0000098), and control cohorts. For case probands, we observed a partial overlap in height growth chart values exceeding the CDC-recommended outlier threshold for short or tall stature (height Z-score ≤ 2 or ≥ 2, respectively). For the short stature cohort (N = 222), 103 (46%) passed the threshold, and for tall stature cohort (N = 35), 20 (57%) passed the corresponding threshold. For probands with observed outlier height Z-scores we found that their trait-relevant PGS liability tended to stratify into expected direction of effect. Specifically, for the short stature cohort with outlier height Z-score (N = 103), 84 (82%) had a PGS ≤ 0 and 19 (18%) > 0. For tall stature (N = 20), 19 (95%) had a PGS ≥ 0 and 1 (5%) <0 (Fig. 4A). We replicated this finding in an external rare disease cohort (Deciphering Developmental Disorders (DDD) (N = 766). For the short stature cohort with outlier height observations (N = 52), 44 (85%) had a PGS ≤ 0 and 8 (15%) > 0. For tall stature (N = 7), all probands had PGS ≥ 0 (Fig. S10). Focusing on the obesity HPO cohort (N = 123), we found that 64 of 95 (67%) probands with a BMI Z-score passing the CDC threshold of obesity had a BMI PGS ≥ 0, and 42 of 52 (81%) with BMI Z-score passing the threshold for severe obesity had a BMI PGS ≥ 0 (Fig. 4B). These findings highlight the potential for enhanced rare disease phenotyping through integrating EHR and PGS information.
Enrichment for rare disease variants in putative core PGS genes
We next assessed the overlap of clinical diagnostic or candidate (VUS) rare disease variants in genes that are also present in significantly associated PGS. Across all PGS genes previously linked to the associated HPO rare disease phenotype in OMIM or Orphanet (HPO gene associations; see Data Availability), we observed a 6-fold increase in cases having a diagnostic or candidate rare disease variant in a PGS gene compared to controls (N = 538; Fisher’s Exact Test (two-sided), odds ratio = 6.23 [CI 5.85–6.63], P < 1 × 10−16) (Fig. 5A). We further implemented a method for ranking PGS genes to define a set of core/key genes where a rare variant has the potential to exert a relatively larger effect on disease risk, as postulated in the omnigenic model of complex traits13. Previous studies have shown that GWAS effect sizes tend to be larger for genes overlapping known disease-matched monogenic disorder genes10; given this finding, we ranked genes in each PGS using summarized effect weights for variants in a PGS that are within or proximal (± 10 Kb) to any protein coding gene (Methods). We observed for case probands an increasing enrichment in overlap in clinical diagnostic or candidate variants in genes with higher rank in the PGS. For example, for the top 0.01% effect rank of PGS genes we observed a 30-fold increase in clinical diagnostic or candidate variant overlap in cases compared to controls (Fisher’s exact test (two-sided), odds ratio = 31.03 [CI 16.04–57.63], P < 1 × 10−16). This effect was stronger when increasing the stringency of HPO-PGS significance (Fisher’s Exact Test (two-sided): FDR 10%: odds ratio = 65.37 [CI 29.01–151.06], P < 1×10−16; FDR 5%: odds ratio = 79.00 [CI 29.33–246.59], P < 1 × 10−16) (Fig. S11A). We observed a similar trend using data from an external rare disease cohort (Deciphering Developmental Disorders (DDD)) (Fig. S11B).

a Enrichment for HPO cases versus controls (total N = 538) with a diagnostic or candidate (VUS) rare disease variant in an associated PGS gene with indicated effect rank percentile in PGS. Dots indicate the odds ratio from logistic regression (two-sided), error bars indicate 95% confidence interval. P-values for PGS gene rank percentile: All, P = 0; 90, P = 3 × 10−127; 95, 5 × 10−89; 99, 1 × 10−45; 99.5, 1 × 10−31; 99.9, 1 × 10−17. No adjustments were made for multiple comparisons. b Proportion of cases (top: obesity cohort; bottom: short stature cohort) and controls with rare variants prioritized in to ACMG severity categories 2 or 3 in trait-relevant eQTS gene as a function of individual liability for significantly associated polygenic scores. P-values are derived from logistic regression (two-sided), testing for an interaction between case/control cohort and PGS Z-score on rare variant burden in indicated eQTS gene set. Ext. denotes maximum outlier PGS cutoff that could be assessed where the cohort contained at least one case with a selected rare variant in an eQTS gene.
Using an orthogonal metric to define putative trait-relevant core/key PGS genes, we focused on trait-matched expression quantitative trait scores (eQTS) genes (eQTLGen Consortium, see Data Availability), currently available for several complex traits and diseases. eQTS measures the correlation between the expression of a given gene and a polygenic score, aggregating regulatory effects on expression for both cis- and trans-acting variants14. The genes with the strongest correlation for a given trait are then defined as putative core/key genes. We assessed the likelihood of a proband harboring a potential large-effect rare variant (ACMG severity category 2 or 3; Methods) in an eQTS gene across increasingly more stringent PGS liability thresholds.
Focusing on a subset of the HPO obesity case/control cohort with clinical sequencing data available (N = 970), we identified candidate rare variants in a set of eQTS genes significantly associated with obesity or extreme body mass index (N genes = 34; Methods). Across the full cohort, 32 of 68 (47%) cases harbored a potential large-effect rare variant in a trait-relevant eQTS gene and 380 of 902 (42%) controls. When we then integrated individual polygenic liability for a significantly associated PGS trait (PGS = “body mass index”) in addition to rare variant burden, we observed an increasing enrichment in the proportion of cases with rare variants in trait-relevant eQTS genes as a function of trait-relevant polygenic liability, but a depletion in controls (logistic regression, interaction P = 3 × 10−03). At an outlier PGS Z-score threshold of ≥ 2, 10 of 14 (71%) cases were found to have a potentially impactful rare variant, and 7 of 24 (29%) controls (Fig. 5B). Increasing the PGS Z-score outlier threshold further (PGS Z-score ≥ 2.5), we observed 5 of 6 (80%) cases with potentially impactful rare variants in trait-relevant eQTS genes and 0 of 6 controls. We repeated this analysis using the same patient cohort across six eQTS gene sets for non-matched complex diseases or traits and observed no significant differences between cases and controls (Figure S12). Notably, of the 34 obesity-associated eQTS genes used in this analysis, only one has been previously linked to a rare Mendelian obesity disorder in OMIM or Orphanet (HPO gene associations; see Data Availability).
We repeated this analysis in a smaller set of available eQTS genes for height (N genes = 10) for the short stature HPO case/control cohort (N = 944), observing a similar interaction between height PGS liability and frequency of rare variants in cases and controls (logistic regression, interaction P = 4 × 10−03). Across the full cohort, 4 of 78 (5%) cases and 26 of 866 (3%) controls harbored a potential large-effect rare variant in a height eQTS gene, and 14% for cases and 0% for controls at PGS Z-score ≤ 2 (Fig. 5B). Again, using eQTS gene sets from non-matched complex diseases or traits, we observed no significance differences between cases and controls (Fig. S13). These findings suggest a potential framework for the discovery of novel rare disease genes integrating rare disease-associated PGS liability, PGS gene effect estimates, and clinical rare variant annotations.
Discussion
Recent studies have shown that integrating PGS information can aid in understanding the genetic basis of rare diseases3,15,16,17,18. Here, we implemented a pipeline for the systematic assessment of PGS associations in a large-scale, phenotypically diverse pediatric rare disease cohort, mapping patient phenotypes to over 500 complex trait PGSs. We demonstrated how individual polygenic liability is enriched in proband carriers of inherited, clinically prioritized variants of unknown significance (VUS) compared to unaffected carrier parents. Furthermore, using two separate metrics to define putative core/key PGS genes, we identified strong overlap between clinical diagnostic or candidate rare disease genes and those with large estimated effects in significantly associated PGS.
The accurate classification of VUS pathogenicity is an ongoing and growing problem in the field19. Our findings provide evidence that integrating information from proband and parent polygenic backgrounds for associated complex traits could be a useful addition to variant annotation workflows. We focused on a set of inherited variants with high clinical suspicion of pathogenicity resulting from an expert clinical geneticist review, suspected to cause disease through autosomal dominance with partial penetrance. An explanation for why some phenotypes comprising the nominated syndrome are not observed in unaffected carrier parents could be helpful in the further annotation of these candidate diagnostic variants. For the set of inherited VUS studied here, observed differences in polygenic background between probands and unaffected carrier parents and siblings might explain variable presentation of the candidate rare disorder, but further work should be conducted using a larger variant set than was available at the time of this study. Additionally, with relevance to ongoing efforts to experimentally catalog the functional impact of VUS and other possible disease variants using multiplexed assays of variant effect (MAVEs) or similar platforms20,21, our results suggest that the utility of these approaches might be maximized when using patient-specific cell lines to reflect potentially relevant polygenic background associated with variable rare disease phenotype presentation. Our findings integrating eQTS annotations to define putative core genes suggests that future diagnostic approaches for rare diseases would benefit from jointly considering patient rare variants and their polygenic liability for associated complex traits.
We partially replicated PGS associations from GA4K using data from an external rare disease cohort comprised of individuals with a known or suspected neurodevelopmental disorder (Deciphering Developmental Disorders (DDD) study). We could attempt to replicate only a subset of HPO phenotypes tested in GA4K, with a bias toward neurodevelopmental disorder-related HPO phenotypes given the composition of DDD. We observed that the frequency of HPO term usage could vary substantially between GA4K and DDD for patients with similar disorders, potentially due to differences in clinical phenotyping protocols. Future improvements for defining the same or similar patient populations might benefit from utilizing the HPO hierarchy to capture more generalized phenotype terms, or in the use of quantitative measures such as metabolites or affected organ morphology.
One key limitation of our approach is the focus on the subset of GA4K samples of European ancestry. This constraint reflects bias toward this ancestry group in available GWAS and PGS training cohorts11, as well as in the GA4K study cohort itself. Although we show high consistency of associated PGS liability across non-European ancestry cohorts available in GA4K, future updates to our work will benefit from ongoing efforts in multi-ancestry PGS development22 and targeted approaches for equitable outreach and enrollment in GA4K23 to increase available non-European cohort sizes. Furthermore, our approach associates single standardized phenotype terms (HPO) with PGS, whereas our results showing a mismatch in growth disorders HPOs and height growth chart Z-scores from EHR data suggest an opportunity for improved rare disease phenotyping through integrating multiple data modalities.
Combined, our study suggests that future advances in the diagnosis of rare diseases will be enabled by considering the full frequency spectrum of genetic variation present in patient cases.
Methods
Ethical declaration
The GA4K study was approved by the Children’s Mercy Institutional Review Board (IRB) (Study # 11120514). Informed written consent was obtained from the parent/guardian of all participants prior to study inclusion. Additionally, assent is obtained from participants if age 7 or older unless the participant has a condition that would limit comprehension.
GA4K genotype imputation
Genotyping of the full available GA4K study cohort was performed using the Avera Global Screening Array (24v1-0_A1, stranded). Genotyping array variant calls in hg38 build for samples with genotyping call rate >= 90% were then used as input for imputation using the TOPMed imputation server with the r3 (1.0.0) reference panel24. The final proband study cohort comprised 1434 female and 1625 male samples.
GA4K ancestry inference
Principal component analysis was performed on a subset of LD and MAF pruned variants from chromosome 1 using plink (version 2)25 (“indep-pairwise” flag, values: 1000, 50, 0.05; “maf” flag, value: 0.01) and flashpcaR (version 2.0.1)26. Using a subset of known ancestry labels, the full GA4K cohort was filtered to define a European (EUR) ancestry cohort used for association analysis comprised of probands with known EUR ancestry and those inferred from manual inspection of a principal components plot (PC1 > 0, PC2 < 0.005).
Filtering PGS and calculating individual scores
Individual-level PGSs for GA4K participants were calculated using PGS weights obtained from The Polygenic Score (PGS) Catalog11 (see Data Availability). From a starting set of 3334 PGS downloaded from PGS Catalog (February 2023), PGS measuring the same trait or disease were filtered by retaining the PGS with the greatest number of variants. Further, PGS measuring lifestyle phenotypes such as diet were removed. PGS containing duplicated variants (more than one weight listed for the same variant) were removed. PGS variants were converted to hg38 coordinates using dbSNP27 (version 155) where necessary. PGS with < 25 variants were removed. 1102 PGS remained after applying these filtering criteria. PGS were linked to category labels using the PGS ontology ID and trait category annotations available from PGS Catalog. PGS variants were restricted to autosomes only. Individual PGSs were calculated using plink (version 1.9)25 (“sum” flag) on all variants available in the GA4K imputed genotype callset with R2 ≥ 0.8 (“exclude-if-info” flag). PGS scores were converted to Z-scores for each PGS in each ancestry group separately. Samples with an extreme outlier PGS Z-score (abs(PGS Z-score)≥6) in one or more PGS, or clear outliers in PCA clustering, were removed from further analysis. Each ancestry cohort was then filtered to include probands only. The remaining samples were retained for analyses using mother, father, and/or sibling PGS data, where indicated.
Identifying significant HPO-PGS associations
PGSs were associated with rare disease case sub-cohorts using HPO IDs for any ID with a minimum proband count 5 among the 2641 proband study cohort (N = 626 sub-cohorts). For each HPO sub-cohort and PGS (N HPO = 626; N PRS = 1102; N = 689,852 pairwise comparisons) a logistic regression model was used with HPO case/control status as the response variable and PGS Z-score, first five principal components of ancestry (see “GA4K inferred ancestry”), and sex as predictor variables. Model fit was assessed with Nagelkerke’s R squared using the NagelkerkeR2 function in the fmsb package (version 0.7.5) in R. An empirical P-value was calculated for each pairwise comparison using a simulated null distribution of logistic regression abs(Z-statistics) across 10,000 permutations of random case/control label reassignment (retaining relative HPO case/control counts) within each HPO-PGS pair (approximately 6.9B total model calculations). Predictor variables remained as described above. Empirical P-values were generated in R using the empPvals function available in the qvalue package (version 2.30.0)28. The empirical P-value is defined here as the fraction of null logistic regression abs(Z-statistics) that match or exceed the observed abs(Z-statistic) (plus an integer constant) for a given HPO-PGS pair. In this way, the minimum possible empirical P-value for a given HPO-PGS pair in the present analysis is 1 × 10−04 (i.e., the observed statistic is not matched or exceeded by any of the 10,000 null statistics from permutations). The expectation of this procedure is that the distribution of null empirical P-values follows a uniform distribution29; however, when combining empirical P-values across all HPO-PGS pairs it was observed that this distribution was not uniform (Kolmogorov-Smirnov test) and that the estimate of null statistics was affected by the relative sample size of each HPO sub-cohort. Therefore, the false discovery rate (FDR) was calculated within each HPO case sub-cohort separately to conform with the expected uniform distribution of empirical P-values.
Linking PGS variants to genes and effect size ranking
PGS variants within or proximal (± 10 Kb) to any protein coding gene were identified using bedtools (version 2.29.1)30 (“window” flag; value: 10,000). Gene coordinates and annotations were downloaded from GENCODE (file: gencode.v26.GRCh38.genes.gtf) (see Data Availability). For each PGS, genes were ranked by effect size by selecting the linked variant (i.e. within the gene body or ± 10 Kb) with the maximum absolute PGS effect weight.
GA4K clinical sequencing and phenotypic data
Clinical whole-exome (WES) or whole-genome (WGS) variant calls, standardized phenotype codes (Human Phenotype Ontology (HPO)31), and diagnostic status were obtained from the GA4K study repository. HPO terms were summarized for visualization where indicated using ontology parent terms specified in HPO (see Data Availability), extracted using the get_ancestors function available in the ontologyIndex package (version 2.11)32 in R.
For the analysis of probands with clinical diagnostic status of variant of unknown significance (VUS), probands were selected if their PGS-associated HPO(s) matched any of the disease phenotypes for candidate syndromes returned by clinical annotation performed by clinical geneticists. This annotation pipeline prioritizes disease variants and summarizes matching phenotypes by aggregating across multiple knowledge bases such as OMIM, ACMG, and ClinVar. VUS cases were restricted to single inherited variants (i.e. VUS is present in one parent), autosomal only, annotated as autosomal dominant with partial penetrance, and where associated PGSs were available for the full trio. Compound heterozygous VUS events were removed. Parent VUS zygosity was obtained from clinical WES/WGS available in the GA4K study repository. Carrier parents were confirmed to be unaffected for the matching VUS phenotype(s) by clinical geneticist chart review.
Height and weight observations, including measure date/time and patient ancestry, were obtained from the electronic medical record at the GA4K study site at Children’s Mercy Hospital. Observations were filtered for “White” ancestry label. The R package cdcanthro (version 0.1.1) was used to calculate age and sex adjusted Z-scores for height and weight using CDC guidelines. Further, BMI Z-scores were calculated using the formula Weight/(Height/100)2. The average (median) Z-score for each measurement was computed for samples with more than one observation time point.
Replication in an external rare disease cohort
External replication analysis used case data from the Deciphering Developmental Disorders (DDD) study (see Data Availability), comprising 571 female and 893 male samples. Genotyping array variant calls (dataset EGAD00010002568) (hg38 genome build) were used as input for imputation using the TOPMed imputation server with r3 (1.0.0) reference panel24. Variants were filtered to those with imputation quality R2 ≥ 0.8 and which were also available in the filtered GA4K imputed callset (N variants = 23,952,791). Probands were subset to those with GBR ancestry using resource file EGAF00008185290 in dataset EGAD00010002568. Probands were further subset for those with ≥ 1 entry in phenotyping (HPO) data file EGAF00002191760. We used FAM file EGAF00008195272 in dataset EGAD00010002568 to check for sibling probands, retaining one proband at random as applicable. PGS were calculated as described above (see “Filtering PGS and calculating individual scores”). Clinical variant annotations were obtained from dataset EGAD00001010137, retaining variants with a clinical annotation (column name: “pathogenicity_clinical”) of “pathogenic”, “likely_pathogenic”, or “uncertain”.
Assessing rare variant burden in large-effect PGS genes
To assess the landscape of rare variants in putative core/key PGS genes (eQTS genes) for obesity, rare variant impact annotations were obtained using Variant Effect Predictor (VEP) (version 98_38)33 and allele frequencies from gnomAD (version 3)34. Rare variants were defined as those with gnomAD MAF < 1% (and including variants absent in gnomAD) and were further categorized using ACMG variant severity guidelines35. Variants annotated in category 2 or 3 were retained. Rare variants in category 2 are defined as: nonsense; disruption of stop; loss of initiation; splice junction; donor/acceptor (AG/GT); frameshift; whole transcript deletion. Category 3 variants are defined as: missense; in-frame in/del including whole exon; intronic or synonymous variant possibly affecting splicing (in polypyrimidine tract, five_prime_exonic, five_prime_flank, three_prime_exonic, five_prime_intronic, three_prime_flank); any variant in a mitochondrial gene.
eQTS genes (FDR < 5%) were obtained from eQTLGen Consortium (see Data Availability), and subset using the trait column for the terms “obesity” or “extreme_bmi” for the obesity HPO analysis, and “height” for the short stature HPO analysis. To create a set of non-relevant traits, the trait column was subset for the terms “asthma”, “celiac disease”, “juvenile idiopathic arthritis”, “primary biliary cirrhosis”, “educational attainment”, or “coronary artery disease”. In this analysis, the obesity HPO case cohort was defined as those with a HPO term for “obesity” (HP:0001513), “increased body weight” (HP:0004324), “overweight” (HP:0025502), “truncal obesity” (HP:0001956), “childhood-onset truncal obesity” (HP:0008915), “abdominal obesity” (HP:0012743), or class I-III obesity (HP:0025499, HP:0025500, HP:0025501) as well as a BMI growth chart Z-score > 2. Controls were defined as any proband without any of these HPO terms, as well as an absolute BMI growth chart Z-score of <1. Cases in the short stature analysis were defined as those having a HPO term for “short stature” (HP:0004322). Controls were defined as any proband without “short stature” or “tall stature” HPOs, as well as an absolute height growth chart Z-score <1.
All statistical analyses were performed in R (version 4.2.1). Figures were generated using ggplot2 (version 3.4.0)36,37.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
GA4K data is available via dbGAP using accession number phs002206.v5.p1 and AnVIL [https://anvilproject.org/data/studies/phs002206]. Raw and processed GA4K data are available under restricted access due to IRB regulations and informed consent limiting access to users studying genetic diseases. Data access is provided by dbGAP for certified investigators with local IRB approval in place. Access approvals are for a period of one year, which can be renewed as necessary. Polygenic scores are available from PGS Catalog at https://www.pgscatalog.org/. Gene annotations are available from GENCODE (version 26) at https://www.gencodegenes.org/human/release_26.html. eQTS genes are available from eQTLGen Consortium at https://www.eqtlgen.org/eqts.html. Human Phenotype Ontology (HPO) in OBO format is available at https://hpo.jax.org/app/data/ontology. HPO phenotype-to-gene annotations are available at https://hpo.jax.org/app/data/annotations. Deciphering Developmental Disorders (DDD) data is available from the European Genome-Phenome Archive at the European Bioinformatics Institute using study accession number EGAS00001000775. Source data are provided with this paper.
Code availability
Custom scripts generated for this study can be obtained at https://github.com/csmail/ga4k_pgs.
References
Cohen, A. S. A. et al. Genomic answers for children: dynamic analyses of >1000 pediatric rare disease genomes. Genet. Med. 24, 1336–1348 (2022).
Cheung, W. A. et al. Direct haplotype-resolved 5-base HiFi sequencing for genome-wide profiling of hypermethylation outliers in a rare disease cohort. Nat. Commun. 14, 3090 (2023).
Niemi, M. E. K. et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271 (2018).
Fahed, A. C. et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 3635 (2020).
Schroeder, P. et al. Rare variant association analysis in 51,256 type 2 diabetes cases and 370,487 controls informs the spectrum of pathogenicity of monogenic diabetes genes. medRxiv (2023) https://doi.org/10.1101/2023.09.28.23296244.
Goodrich, J. K. et al. Determinants of penetrance and variable expressivity in monogenic metabolic conditions across 77,184 exomes. Nat. Commun. 12, 3505 (2021).
Chami, N., Preuss, M., Walker, R. W., Moscati, A. & Loos, R. J. F. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med 17, e1003196 (2020).
Davies, R. W. et al. Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat. Med. 26, 1912–1918 (2020).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231.e11 (2020).
Freund, M. K. et al. Phenotype-specific enrichment of mendelian disorder genes near GWAS regions across 62 complex traits. Am. J. Hum. Genet. 103, 535–552 (2018).
Lambert, S. A. et al. The polygenic score catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53, 420–425 (2021).
Oetjens, M. T., Kelly, M. A., Sturm A. C., Martin C. L. & Ledbetter D. H. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun. 10, 4897 (2019).
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034.e6 (2019).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Wigdor, E. M. et al. The female protective effect against autism spectrum disorder. Cell Genom. 2, 100134 (2022).
Michaud, V. et al. The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism. Nat. Commun. 13, 3939 (2022).
Alver, M. et al. Contribution of schizophrenia polygenic burden to longitudinal phenotypic variance in 22q11.2 deletion syndrome. Mol. Psychiatry 27, 4191–4200 (2022).
Škorić-Milosavljević, D. et al. Common genetic variants contribute to risk of transposition of the great arteries. Circ. Res. 130, 166–180 (2022).
Starita, L. M. et al. Variant interpretation: functional assays to the rescue. Am. J. Hum. Genet. 101, 315–325 (2017).
Radford, E. J. et al. Saturation genome editing of DDX3X clarifies pathogenicity of germline and somatic variation. Nat. Commun. 14, 7702 (2023).
Findlay, G. M. et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018).
Fatumo, S. et al. A roadmap to increase diversity in genomic studies. Nat. Med. 28, 243–250 (2022).
Kane, N. J. et al. Committing to genomic answers for all kids: evaluating inequity in genomic research enrollment. Genet. Med. 25, 100895 (2023).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Abraham, G., Qiu, Y. & Inouye, M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 33, 2776–2778 (2017).
Sherry, S. T., Ward, M. & Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9, 677–679 (1999).
Storey, J. D., Xiao, W., Leek, J. T., Tompkins, R. G. & Davis, R. W. Significance analysis of time course microarray experiments. Proc. Natl Acad. Sci. Usa. 102, 12837–12842 (2005).
Storey, J. D., Taylor, J. E. & Siegmund, D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. R. Stat. Soc. Ser. B Stat. Methodol. 66, 187–205 (2003).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Köhler, S. et al. The human phenotype ontology in 2021. Nucleic Acids Res 49, D1207–D1217 (2021).
Greene, D., Richardson, S. & Turro, E. ontologyX: a suite of R packages for working with ontological data. Bioinformatics 33, 1104–1106 (2017).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Chen, S. et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022.03.20.485034 https://doi.org/10.1101/2022.03.20.485034 (2022).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer International Publishing, 2016).
Smail, C. et al. GA4K PGS manuscript code release, https://doi.org/10.5281/zenodo.13307331 (2024).
Acknowledgements
C.S. is supported by NIH grants R35GM146966 and R21HG012422. The work of GA4K was made possible in part by generous gifts to Children’s Mercy Research Institute at Children’s Mercy Kansas City. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute (grant number WT098051). The views expressed in this publication are those of the authors and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
Author information
Authors and Affiliations
Consortia
Contributions
C.S. performed main data analyses and processing; B.G. performed genotype imputation and polygenic score data processing; C.B. performed clinical phenotyping; M.R.K.K, W.A.C. and J.J.J. performed rare variant annotation; C.S.S. performed genotype data processing; K.F. extracted and processed clinical observation data; A.S.A.C., E.G.F., I.T. and C.B. provided clinical variant and diagnostic annotations; E.G. assisted with data interpretation; C.S. wrote the manuscript; C.S., T.P. and E.G. edited the manuscript; C.S. and T.P. conceived of and oversaw the study.
Corresponding authors
Ethics declarations
Competing interests
The Authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Carrie Bearden, Eleanor Seaby and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Smail, C., Ge, B., Keever-Keigher, M.R. et al. Complex trait associations in rare diseases and impacts on Mendelian variant interpretation. Nat Commun 15, 8196 (2024). https://doi.org/10.1038/s41467-024-52407-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52407-1
- Springer Nature Limited