

Abstract
Plasmodium falciparum is the causative agent of malaria and remains a pathogen of global importance. Asexual blood stage replication, via a process called schizogony, is an important target for the development of new antimalarials. Here we use ultrastructure-expansion microscopy to probe the organisation of the chromosome-capturing kinetochores in relation to the mitotic spindle, the centriolar plaque, the centromeres and the apical organelles during schizont development. Conditional disruption of the kinetochore components, PfNDC80 and PfNuf2, is associated with aberrant mitotic spindle organisation, disruption of the centromere marker, CENH3 and impaired karyokinesis. Surprisingly, kinetochore disruption also leads to disengagement of the centrosome equivalent from the nuclear envelope. Severing the connection between the nucleus and the apical complex leads to the formation of merozoites lacking nuclei. Here, we show that correct assembly of the kinetochore/spindle complex plays a previously unrecognised role in positioning the nascent apical complex in developing P. falciparum merozoites.
Similar content being viewed by others


Introduction
Malaria is a mosquito-borne disease that causes devastating health problems. Despite substantial efforts over the last decade to reduce the burden of malaria, this disease still caused an estimated 247 million infections, and claimed 619,000 lives, in 20211. The emergence of malaria parasites resistant to the front-line artemisinin-based antimalarials2, combined with the recent impacts of the COVID-19 pandemic on malaria control efforts3, makes the need to develop new antimalarial strategies even more pressing.
The process of mitosis in P. falciparum differs from that of mammalian cells. Apicomplexan parasites undergo closed mitosis, similar to many fungi, where the nuclear membrane remains intact through the entirety of mitosis4,5. During the intraerythrocytic asexual stage, malaria parasites replicate by a process called schizogony, where several asynchronous rounds of mitosis and karyokinesis (nuclear division) are followed by one round of synchronised karyokinesis and cytokinesis to form a segmented schizont, resulting in the formation of up to 32 daughter merozoites5,6.
In many eukaryotic cells, the centrosome is the microtubule organising centre (MTOC) for the chromatid-separating spindle microtubules. In mammalian cells, the centrosome contains a pair of centrioles, surrounded by pericentriolar material. During mitosis, the centrosome duplicates and the nuclear membrane is disassembled7. Mitotic spindle microtubules are nucleated from the centrosome and extend toward the centre of the cell to capture structures called kinetochores, which, in turn, segregate sister chromatids8.
By contrast, P. falciparum has no canonical centrioles, nor any other distinct structure within the centrosome equivalent, which is termed the centriolar plaque. Early electron microscopy (EM) studies revealed an amorphous structure embedded in the nuclear membrane, with electron-dense signals detected on both sides of the membrane9,10,11,12. The molecular composition and architecture of the Plasmodium centriolar plaque remain poorly understood. A limited number of canonical centrosome components have been identified. These include centrins—structural proteins that belong to a small family of calcium-binding proteins with four EF-hand domains13,14,15,16,17. A Sfi1-like protein (PfSlp)18 and a homologue of the Aurora kinase (PfArk1)19 have also been identified. Each of these proteins is associated with the outer centriolar plaque region.
3D-reconstruction electron microscopy of the mitotic spindle of P. falciparum schizonts revealed 14 pairs of kinetochores12. Many kinetochore proteins have been determined in P. berghei and the related apicomplexan parasite, Toxoplasma gondii20,21,22,23. A key component of the kinetochore is a tetrameric complex that comprises heterodimers of Nuclear Division Cycle 80 protein (NDC80) and Nuf2, with heterodimers of Spindle Pole Component (SPC)-24 and SPC25. The C-termini of the SPC24/SPC25 heterodimer provide a link to CENH3 (CENP-A) a component of the centromeres, while the NDC80-Nuf2 heterodimer interacts with the spindle microtubules22,24,25,26.
In mammalian cells, CENP-A depletion leads to destabilisation of the kinetochore complex, which, in turn, compromises spindle microtubule stability, resulting in release of microtubule minus-ends from the centrosome and dispersal of the pericentriolar material27. These data suggest a functional interdependence of the centromeres and the centrosomes during mitosis27.
NDC80 and Nuf2 have been studied in P. berghei blood stages using live cell reporters, and are first detected in the trophozoite stage, as mitosis begins23,28. In segmented schizonts, which have completed mitosis, the NDC80 signal is still detected, while the Nuf2 signal is not23,28. Previous studies showed that NDC80 can distribute along the nuclear microtubules in Plasmodium17,20, in contrast to mammalian kinetochores, which bind predominantly at the plus ends. Efforts to knockout or knockdown NDC80 and Nuf2 in P. berghei were not successful, consistent with these genes being essential20,23. Similarly, knock sideways and promoter swap methods in P. falciparum and P. yoelii confirm that NDC80 plays an essential role in replication28,29,30.
In this work, we generate transfectants expressing tagged versions of the kinetochore proteins, PfNDC80 and PfNuf2, under conditional knockout/knockdown control. Combining classical cell and molecular biology techniques with live cell imaging, Ultrastructure-Expansion Microscopy (U-ExM) and electron microscopy, we demonstrate that PfNDC80 and PfNuf2 play essential and unexpected roles in asexual blood stage proliferation. Deletion of PfNDC80 and PfNuf2 leads to loss of the signal for the centromere marker, CENH3, suggesting disruption of the centromere complex. It also breaks the nexus between the spindle microtubules and the nuclear membrane-embedded centriolar plaque/MTOC, leading to disorganised spindles and uncoupling of the nucleus from the apical complex. The centrin-containing outer centriolar plaque disengages from the nucleus, but still directs assembly of sub-pellicular microtubules and apical organelles, leading to packaging of anucleate merozoites.
Results
PfNDC80 and PfNuf2 undergo dynamic reorganisation
We targeted the native locus of the kinetochore components, PfNDC80 (PF3D7_0616200) and PfNuf2 (PF3D7_0316500), using a CRISPR-Cas9 homology driven approach, incorporating a 3×HA tag and a glmS ribozyme sequence into the 3′ end of the loci. LoxP sites were introduced downstream of the HA tag via the insertion of an artificial intron within the coding region (Supplementary Fig. 1a). Introduction of loxP and glmS ribozyme sequences allows for conditional knockout (cKO) of the genes and knockdown (cKD) of the transcripts, respectively31,32 (Supplementary Fig. 1a), which ensures very efficient and sustained knockdown of the gene product. Integration into the endogenous loci and excision progress of target gene were monitored at different time points by PCR (Supplementary Fig. 1b, c). The level of expression of the HA-tagged proteins was monitored by Western blotting (Supplementary Fig. 1d, e). Wide-field immunofluorescence microscopy revealed PfNDC80 and PfNuf2 as punctate structures within the nuclei (Supplementary Fig. 1f, g).
Following validation of the cell lines, we examined the location of the two kinetochore components at different stages of mitosis (Fig. 1a). U-ExM has recently been applied to visualise a range of cellular processes in P. falciparum16,33,34,35,36,37. Here, the PfNDC80 and PfNuf2 transfectant-infected red blood cells (RBCs) were embedded, and gels were expanded 4.5 times (on average), labelled, mounted, and imaged by Airyscan confocal microscopy. NHS ester-conjugated, sulfonated fluorophores are water-soluble probes that react with primary amines in proteins and other molecules to yield stable amide bonds. Suspending DyLight™ 488-NHS ester in phosphate-buffered saline (PBS) (NHS-PBS) gives good definition of protein-rich structures, such as the rhoptries37 (Fig. 1b, right-hand side, depicted in greyscale/inverse greyscale). Here, we explored a different method for general protein labelling, which yields improved membrane labelling. We found that applying DyLight™ 488-NHS ester suspended in 3% bovine serum albumin (NHS-BSA) gives good delineation of the RBC membrane, the parasite plasma membrane (PPM) and nuclear membrane (Fig. 1b, left-hand side, depicted in greyscale/green). We used both protocols in this study, with the NHS-PBS fluorescence signal depicted in inverted greyscale and the NHS-BSA signal in green. The HA-tagged PfNDC80 and PfNuf2 structures were visualised using anti-HA antibodies. Progress through blood stage mitosis is readily tracked by labelling microtubule structures with anti-β-tubulin or anti-α-tubulin and the chromatin with DAPI.

a Illustration of P. falciparum mitotic stages. The diagrams represent parasites with a hemispindle, a mitotic spindle, an interpolar spindle, a spindle remnant and a formed merozoite. The apical complex is assembled above a nuclear membrane-embedded centriolar plaque, with inner and outer regions. Structures are labelled in the diagrams or defined in the key at the bottom of the diagram. b U-ExM images of schizont-infected RBCs labelled with DyLight488-NHS-ester in BSA (NHS-BSA, greyscale and green) or DyLight488-NHS-ester in PBS (NHS-PBS, greyscale, and inverted greyscale). NHS-BSA (left panels) gives good labelling of membranes. The nuclear membranes are highlighted. NHS-PBS (right panels; with zoom inset) gives good labelling of protein-rich structures, such as the rhoptries and centriolar plaque. U-ExM images of (c) PfNDC80-HA transfectants (anti-HA (PfNDC80), greyscale, yellow arrowheads), counter-stained with NHS-BSA (green) and (d) PfNuf2-HA/GFP-PfCENH3 transfectants (anti-HA (PfNuf2), green, and anti-GFP (PfCENH3); red, yellow arrowheads) counter-stained with NHS-PBS (greyscale), revealing the kinetochore and centromere components co-located and relocating from the base of the hemispindles to the mid-plane of the mitotic spindle, then contracting to the nuclear periphery. The images are displayed as z-projections. Anti-β-tubulin (anti-β-tub, red) marks the spindle microtubules (intranuclear) and sub-pellicular microtubules (SP-MT, white arrowheads). DAPI (blue) stains the chromatin. Zoom-out/zoom-in scale bars are 5 μm and 2 μm. Additional images/ channels are presented in Supplementary Fig. 2 (PfNDC80-HA), Supplementary Fig. 3 (PfNuf2-HA) and Supplementary Fig. 5a (PfNuf2-HA/ GFP-PfCENH3) and 5b (PfNDC80-HA/ GFP- PfCENH3). Figure 1c, d experiments were performed 3 times from 3 different sample preparations.
The dynamic organisation of kinetochore proteins, Nuf2, NDC80 and Apicomplexan Kinetochore protein1 (AKiT1), has previously been studied in P. berghei5,20,23,38,39. Consistent with those reports, in nuclei preparing for karyokinesis, the PfNDC80 and PfNuf2 signals (Fig. 1c and Supplementary Figs. 2a, b, 3a, b, first rows, yellow arrowheads) are concentrated near the base of anti-β-tubulin-labelled hemispindles, i.e., at the periphery of the nucleus, as marked by DAPI (blue) and the NHS-BSA-labelled membrane (green) (Fig. 1c and Supplementary Figs. 2a, 3a). During nuclear division, PfNDC80 and PfNuf2 signals (Fig. 1c and Supplementary Figs. 2a, b, 3a, b, second row, yellow arrowheads) are often observed as a pair of parallel lines at the mid-plane of the mitotic spindle. In some cells, extended spindles, stretching between separating nuclei, are observed (Fig. 1c, second row, zoom 1, yellow arrowheads), with the kinetochore marker concentrated towards the spindle origins. In segmenting schizonts (Fig. 1c and Supplementary Figs. 2a, b, 3a, b, third row), PfNDC80 and PfNuf2 (yellow arrowheads) are associated with the contracted spindle remnant. Nascent sub-pellicular microtubules (white arrowheads) are evident just above the spindle remnant, as reported previously5,38,39. In segmented schizonts (Fig. 1c and Supplementary Figs. 2a, b, 3a, b, fourth row), the kinetochore components (yellow arrowheads) contract back to a location at the nuclear periphery, and the sub-pellicular microtubules (white arrowheads) are evident outside the nucleus under the PPM, consistent with previous reports16,20,23,35. The NHS-PBS counterstained samples (Supplementary Figs. 2b, 3b), reveal that the kinetochore marker is associated with protein-rich structures that migrate from the nuclear periphery to the spindle midplane and back again.
Kinetochore and centromere markers are co-located during nuclear division
To investigate the centromere location relative to PfNDC80 and PfNuf2, we used a previously generated a GFP-PfCENH3 (PF3D7_1333700) reporter construct17 and transfected it into the PfNDC80-HA and PfNuf2-HA lines (Supplementary Fig. 4). Figure 1d illustrates the co-location of the centromere (PfCENH3, green) and kinetochore (PfNuf2, red) markers. Prior to nuclear division, the kinetochores and centromeres are sequestered to the strongly labelled NHS-PBS inner centriolar plaque region (Fig. 1d and Supplementary Fig. 5a, top row, yellow arrowhead). As the centriolar plaque duplicates and separates, the kinetochore (PfNuf2, red) and centromere (PfCENH3, green) markers migrate in a pattern that overlaps with the NHS-PBS bright material (Fig. 1d and Supplementary Fig. 5a, second row, yellow arrowheads). An equivalent pattern is observed for PfNDC80-HA and GFP-PfCENH3 (Supplementary Fig. 5b.) The data suggest that the NHS-PBS bright, inner centriolar plaque is comprised of sequestered kinetochore and centromere complexes.
Reorganisation of the centriolar plaque, kinetochore and apical complex
We used a monoclonal anti-centrin antibody to probe the reorganisation of the centriolar plaque at different stages of nuclear division. Early in division the centrin-labelled punctum is located on the cytoplasmic side of the nuclear membrane (Supplementary Fig. 6a; yellow arrowheads), directly opposite the site of formation of the hemispindle (green microtubules) (Supplementary Fig. 6a). Following completion of nuclear division, the spindle microtubules contract to stubs (green signal, aqua arrowheads) that sits just below the centrin punctum (yellow arrowheads) (Supplementary Fig. 6c). At this stage, short stubs of cytoplasmic microtubules (white arrowheads) are observed, apparently arising from the centrin-labelled punctum. Additional examples of each step described above are provided in Supplementary Fig. 7a–c. These data are consistent with previously published data37.
Developing rhoptries are evident as very strongly NHS-PBS-labelled bulbous structures located towards the outside of the cell (Supplementary Figs. 6b-d,7b, c, 8 purple arrowheads). In earlier rounds of nuclear division, each nucleus is associated with one rhoptry (Supplementary Figs. 6b, 7b, first and second row), increasing to two rhoptries in the final round of nuclear division (Supplementary Figs. 6c, 7b, third and fourth row). A continuum of NHS-PBS-labelling is observed from the rhoptries to the centrin-labelled puncta that lie at the surface of the nucleus (Supplementary Figs. 6b-d, 7b, 8), as reported previously17,35.
In NHS-BSA-labelled schizonts that are approaching maturity, anti-β-tubulin-labelled sub-pellicular microtubules (Supplementary Fig. 6e, aqua arrowheads) are evident outside the nucleus, directly above the puncta of retracted spindle microtubules (Supplementary Fig. 6e, yellow arrowheads). Subpellicular microtubules are partially polyglutamylated, and one end of these apically located microtubule structures is recognised by anti-polyE antisera, confirming they are subpellicular microtubules (Supplementary Fig. 6e, orange arrowheads), as previously described17,33. Anti-PfGAP45 (greyscale), a marker of the inner membrane complex (IMC)40, is evident as a cap above each nucleus (Supplementary Fig. 6f, magenta arrowheads). The sub-pellicular microtubules (aqua arrowheads) lie under the IMC, close to the intra-nuclear spindle remnants (Supplementary Fig. 6f, yellow arrowheads). Additional examples are presented in Supplementary Fig. 9a (anti-polyE) and Supplementary Fig. 9b (anti-PfGAP45).
Taken together with previous reports17,35, these data indicate connections from the spindle and inner centriolar plaque through to the outer centriolar plaque, the nascent cytoplasmic microtubules, the apical complex, and the IMC and the PPM. The data show that the spindle microtubules are initiated from a protein-dense structure inside the nucleus, that contains the kinetochore proteins, PfNuf2 and PfNDC80. In addition, we show that cytoplasmic microtubules nucleate close to the outer centrin-containing plaque, corresponding to either the centriolar plaque itself or a closely apposed nascent apical polar ring.
PfNDC80 deletion impacts spindle microtubules and centromeres
We subjected PfNDC80-HA-diCRE-glmS transfectants to rapamycin/ glucosamine treatment for 41 h (initiated at 0–2 h post-invasion (hpi)). This dual conditional knockout and knockdown approach is abbreviated as cKO/KD. PCR and Western analysis show excision and almost complete loss of protein by 30 hpi (Supplementary Fig. 1b, d). As anticipated, cKO/KD of PfNDC80 severely impacts the survival of the blood stage parasites with parasites unable to complete schizogony (Fig. 2a). To investigate the defect more closely, we examined the physical organisation of the mitotic machinery in tightly synchronised early- and mid-stage mitotic schizonts (at 30 and 36 hpi), labelled with anti-β-tubulin and DAPI and imaged using U-ExM (Fig. 2b, c). In control cells, hemispindles, mitotic spindles and extended spindles are observed (Fig. 2b, c, control, yellow arrowheads). In PfNDC80-cKO/KD transfectants, the spindle microtubules show a disrupted organisation. In early-stage cells with a single nucleus (Fig. 2b, cKO/KD, fourth row, yellow arrowheads), elongated microtubules are evident that appear to have been separated from the bulk nuclear material. As nuclear division progresses, microtubules continue to form in the nucleus but fail to converge at an MTOC at the nuclear periphery (Fig. 2b, cKO/KD, fourth and sixth row, yellow arrowheads); and typical hemispindle and mitotic spindle structures are absent (Fig. 2b, c, cKO/KD, yellow arrowheads). Abnormal microtubules were observed in all anti-HA-negative PfNDC80-HA cKO/KD cells examined at 36 and 38 hpi (Fig. 2d).

a Assay of asexual growth of PfNDC80-HA parasites following conditional knockout/knockdown (cKO/KD). Parasites were treated with rapamycin and glucosamine (cKO/KD) or DMSO (control) and the parasitemia was monitored for 4 cycles by flow cytometry. The experiment was performed 3 times, with means and standard deviations presented. Representative U-ExM images of control and PfNDC80-cKO/KD schizonts during early (b 30 hpi) and mid (c 36 hpi) rounds of nuclear division. PfNDC80-HA is visualised using anti-HA (green, b). The spindle microtubules (yellow arrowheads) were labelled with anti-β-tubulin (red, anti-β-tub), chromatin was probed with DAPI (blue), and NHS-PBS (inverse grayscale, b) was used to label the general proteins while the NHS-BSA (green, c) was used to enhance membrane labelling. The images are displayed as z-projections. All the image scale bars are 5 μm, except the zoom images, which are 2 μm. Figure 2b experiments were performed 1 time and Fig. 2c experiments were performed 3 times. d Analysis of the percentage of normal spindle microtubules in anti-HA-positive control and anti-HA-negative cKO/KD PfNDC80-HA cells (36 and 38 hpi). Control, n = 20; cKO/KD, n = 20. The images were analysed from 3 independent experiments. The mean and standard deviation are plotted. Individual data points are shown. Statistical differences were determined using an unpaired Mann–Whitney t-test (****p < 0.0001).
We next examined the reorganisation of the centromere marker, PfCENH3, following cKO/KD of PfNDC80. As described above, in control cells PfNDC80-HA (red) and GFP-PfCENH3 (green) are co-located during chromatid duplication and separation (Fig. 3a, yellow arrowheads, control). As expected, disruption of PfNDC80 leads to loss of the red fluorescence signal (Fig. 3a, cKO/KD). Interestingly, the signal for GFP-PfCENH3 (green) also disappeared in anti-HA-negative PfNDC80-HA/GFP-PfCENH3 cells (Fig. 3a, b, cKO/KD). This suggests that sequestration of PfCENH3 into kinetochore/centromere complexes is lost and that PfCENH3 either disperses throughout the nucleus or is unstable and becomes degraded. Individual channels and additional examples of early, mid, late and segmented schizonts (control and cKO/KD) are presented in Supplementary Fig. 10.

a Representative U-ExM micrographs of PfNDC80-HA/GFP-PfCENH3 control and conditional knockout/knockdown (cKO/KD) mid stage schizonts counterstained with the general protein dye NHS-PBS (inverse grayscale). The anti-HA (PfNDC80-HA, red) labelling overlaps with the anti-GFP (GFP-PfCENH3, green) labelling (yellow arrowheads). In cKO/KD cells, NHS-PBS-labelled apical complex (aqua arrowheads) is separated from the DAPI-labelled nucleus (blue). The anti-GFP signal is also lost. Additional images are presented in Supplementary Fig. 10. The experiment was performed 3 times. b Analysis of the percentage of centriolar plaque structures (labelled with NHS-PBS) associating with anti-GFP puncta in anti-HA-positive control and anti-HA-negative cKO/KD PfNDC80-HA/GFP-PfCEH3 cells (34 and 36 hpi). Control, n = 20; cKO/KD, n = 20. The mean and standard deviation are plotted. Individual data points are shown. An unpaired Mann–Whitney t-test was performed (****p < 0.0001). The images for quantifications were acquired from 3 independent experiments. Figure 3a, b experiments were repeated 3 times. c In late stage schizonts (38 hpi), anti-centrin labels the outer centriolar plaque (red, orange arrowheads), while anti-α-tubulin (greyscale, anti-α-tub) labels the sub-pellicular (aqua arrowheads) and spindle remnant (yellow arrowheads) microtubules. Additional images are presented in Supplementary Fig. 11a, b. The experiment was performed two times. d Analysis of centrin puncta positions. Control, n = 20; cKO/KD, n = 20. The mean and standard deviation are plotted. Individual data points are shown. An unpaired Welch’s t-test was performed to test significance (****p < 0.0001). The images for analysis were acquired from 2 independent experiments. Figure 3c, d experiments were repeated 2 times. e Late stage mitotic schizonts (38 hpi). The sub-pellicular microtubules (aqua arrowheads) are distinguished by labelling with anti-β-tubulin (red, anti-β-tub) and anti-polyE (greyscale). The experiment was performed one time. f The inner membrane complex is labelled with anti-PfGAP45 (greyscale, magenta arrowheads, anti-GAP45). The post-mitotic spindle remnant, inside the nucleus, is labelled with anti-β-tubulin (red, yellow arrowheads). Additional images are presented in Supplementary Fig. 11c, d. The experiment was performed twice. The images are displayed as z-projections. All the image scale bars are 5 μm, except the zoom images, which are 2 μm.
Deletion of PfNDC80 impacts merozoite formation and cytokinesis
We investigated if PfNDC80 cKO/KD affects the final round of nuclear division (38–40 hpi) when the cell initiates cytokinesis and the nuclei are packaged into forming merozoites. In control cells, the spindle remnant (Fig. 3c, control, yellow arrowheads) is positioned towards the outside of the parent cell, close to the centrin punctum (control, orange arrowheads). Upon disruption of PfNDC80, severe disorganisation of the schizont is observed. The spindle microtubule remnant is located away from the nuclear periphery (Fig. 3c, cKO/KD, Zoom 1, yellow arrowheads), while the centrin puncta are aggregated in a region of the cell away from the nuclei (Fig. 3c, cKO/KD, Zoom 2, orange arrowheads). Quantitation reveals that in controls, all the centrin puncta are located just outside of the nuclear membrane, while in contrast only 18% of the centrin puncta are located correctly following cKO/KD of PfNDC80 (Fig. 3d). In control cells, the nascent sub-pellicular microtubules, marked by anti-polyE (Fig. 3e, aqua arrowheads), are evident as narrow structures that are located under the IMC, marked by anti-PfGAP45 (Fig. 3f, magenta arrowheads). In the cKO/KD parasites, the sub-pellicular microtubule/IMC complex still forms and is located close to the centrin puncta, but is distal to the nuclei (Fig. 3c, e, f, cKO/KD). Additional examples and individual channels are presented in Supplementary Fig. 11a, b (anti-centrin), c (anti-polyE), d (anti-PfGAP45).
PfNDC80 is required for correct schizont segmentation
We next imaged fully segmented schizonts to visualise the impact of PfNDC80 cKO/KD on merozoite formation. In control cells, the NHS-BSA signal reveals fully formed merozoites, each containing a compact nucleus located at the basal end of the merozoite (Fig. 4a, b, control). The PfGAP45-labelled IMC forms a sheath around each of the merozoites in the segmented schizont (Fig. 4a, magenta arrowheads). The polyE labelled sub-pellicular microtubules radiate from the apical end of the merozoite (Fig. 4b, aqua arrowheads).

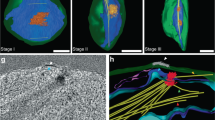
a, b Representative U-ExM images of segmented schizonts in control (DMSO) and PfNDC80-cKO/KD (conditional knockout/knockdown, treated with rapamycin and glucosamine) parasites. The sub-pellicular microtubules are labelled with anti-β-tubulin (anti-β-tub, red, aqua arrowheads) and the chromatin with DAPI (blue). NHS-BSA (green) counterstains the membranes. a Anti-PfGAP45 labels the inner membrane complex (anti-GAP45, greyscale, magenta arrowheads). The experiments were performed twice. b Anti-polyE labels the sub-pellicular microtubules at their minus ends (greyscale, aqua arrowheads). The experiment was performed one time. All images are presented as z-projections. All scale bars are 5 μm, except the zoom images which are 2 μm. Additional images are presented in Supplementary Fig. 12a, b. c, d. Quantification of nuclei parameters for control and PfNDC80-cKO/KD segmented schizonts based on U-ExM data (n = 24 cells). The fraction of unpackaged nuclei c and akaryotic merozoites d are presented (n = 24 cells). Statistical differences were determined using an unpaired Welch’s t-test for c and d (****p < 0.0001). Further quantitative analysis is presented in Supplementary Fig. 12c–f. Figure 4c, d images for analysis were acquired from three independent experiments. e Live cell imaging of control and PfNDC80-cKO/KD segmented schizonts before (upper row) and after (lower row) egress. The subpellicular microtubules were labelled with Tubulin Tracker (TT, red) and chromatin was labelled with Hoechst (greyscale). The yellow arrowheads indicate akaryotic merozoites and blue arrowheads indicate naked nuclei. All images are presented as z-projections. The scale bar is 2 μm. The experiments were performed 3 times. f Single section electron micrographs of segmented schizonts in control and PfNDC80-cKO/KD parasites. The nuclei (N) are marked and the rhoptries are indicated with yellow arrowheads. g 3-D array tomography reconstructions of segmented schizonts in control and PfNDC80-cKO/KD parasites. Figure 5f, g experiments were performed one time. Colour legend: Green, nucleated daughter merozoite plasma membrane; red, anucleate daughter merozoite plasma membrane; blue, nuclei; yellow, rhoptries. f Scale bar is 1 μm.
In PfNDC80 cKO/KD parasites, the apical structures, delineated by the IMC marker (PfGAP45) and polyE-labelled subpellicular microtubules, accumulate in one region of the parent cell. The apical complexes are physically separated from the nuclei, which accumulate in another region of the schizont (Fig. 4a, b, cKO/KD, Supplementary Fig. 12a, b, cKO/KD). Quantitative analysis of control and PfNDC80 cKO/KD parasites revealed that 66% of nuclei are unpackaged (i.e., DAPI-positive, PfGAP45-negative) (Fig. 4c), and 58% of merozoites are nucleus-free (i.e., PfGAP45-positive but DAPI-negative) (Fig. 4d, Supplementary Fig. 12c, d). We also analysed the ratio of the number of merozoites to the number of nuclei per cell. In controls, there were equal numbers of nuclei and apical complexes (i.e., ratio of 1), while in the PfNDC80 cKO/KD parasites the ratio was 0.8, suggesting a partial defect in nuclear division in addition to the nucleus capture defect (Supplementary Fig. 12e). Closer examination of the images suggests inconsistency in the size of the individual nuclei being produced in the PfNDC80-cKO/KD parasites. Quantification reveals a significant decrease in the mean (expanded) volume of the nuclei of cKO/KD parasites (37 ± 34 μm3) compared with controls (43 ± 10 μm3), as well as the presence of some very large volume nuclei (up to 200 μm3), consistent with uneven inheritance of the nuclear contents (Supplementary Fig. 12f).
Live cell imaging of control PfNDC80-tagged mature schizonts and egressing merozoites (41 hpi) reveals fully segregated nuclei, with Tubulin-Tracker-labelled (TT, red) subpellicular microtubules adjacent to the nuclei (Fig. 4e, control). In PfNDC80-cKO/KD schizonts (Fig. 4e, cKO/KD), the nuclei (aqua arrowheads) are segregated away from the subpellicular microtubules. Interestingly, PfNDC80-cKO/KD schizonts still appear to undergo egress as evidenced by differential interference contrast (DIC) imaging. The daughter merozoites appear to lack nuclei (Fig. 4e, yellow arrowheads). Taken together, our results demonstrate that PfNDC80 loss is associated with a defect in segmentation of schizonts.
Electron microscopy analysis confirms the mislocation of the nuclei upon PfNDC80 cKO/KD
To investigate the ultrastructural morphology of the PfNDC80-cKO/KD parasites at the nanoscale, we performed thin section transmission electron microscopy of segmented schizonts (Fig. 4f). In control cells, newly formed merozoites are evident. The electron dense apical organelles, e.g., rhoptries (yellow arrowheads), are positioned at one end of the merozoite, while the nucleus is observed in the basal region (Fig. 4f, control). This arrangement can be clearly visualised in 3D-rendered Array Tomography data showing the rhoptries (yellow) locating at the apical end and the nuclei (blue) at the basal end of the merozoites (membranes in green) (Fig. 4g, control, see Video S1). In contrast, in the PfNDC80-cKO/KD schizonts, the nuclei are aggregated to one side of the cell. Merozoites that lack nuclei but contain rhoptries (yellow arrows) and regions of IMC (observed as membrane thickening), are clustered on the other side of the schizont (Fig. 4f, cKO/KD). This is readily visualised in the Array Tomography data (Fig. 4g, cKO/KD), and associated 3D rotations (Video S2), where the akaryotic merozoites and nuclei are observed on opposing sides of the schizont. These data confirm the live cell and U-ExM data showing mis-segregation of the nuclei and akaryotic merozoites in PfNDC80-cKO/KD schizonts.
PfNuf2 is also essential for normal karyokinesis and cytokinesis
We next investigated the function of a second kinetochore localising protein, PfNuf2. cKO/KD of the PfNuf2-HA-diCRE-glmS locus was confirmed by PCR and Western blotting, showing an almost complete gene excision and loss of protein by 30 hpi (Supplementary Fig. 1c, e). As anticipated, disruption of PfNuf2 seriously impacts parasite replication, with a 90% reduction in growth, when compared to the controls (Supplementary Fig. 13a).
PfNuf2-cKO/KD parasites exhibit a similar phenotype to PfNDC80-cKO/KD parasites. In early nuclear division (30 hpi), NHS-PBS-labelled structures are observed associated with aberrant spindles microtubules or with microtubules that are apparently disconnected from the nucleus (Fig. 5a, cKO/KD, second row, aqua arrowheads, Supplementary Fig. 13b, cKO/KD, yellow arrowheads). In mid mitosis during karyokinesis, dividing nuclei harbouring aberrant or no spindle microtubules are observed (Fig. 5a, fourth row, 5b, cKO/KD; Supplementary Fig. 13c, cKO/KD, yellow arrowheads). In late and segmented schizonts, disconnected sub-pellicular microtubules are observed (Supplementary Fig. 13d, e, cKO/KD, aqua arrowheads). As observed for PfNDC80 cKO/KD, the PfCENH3 signal is lost following cKO/KD of PfNUF2, indicating disruption of the centromeres (Fig. 5c, cKO/KD). Detailed examination of these NHS-PBS-labelled structures in the PfNuf2-cKO/KD parasites shows that the outer centriolar plaque and nascent apical complex are still generated but have lost the connection to the nucleus and are instead located close to the PPM (diagram in Fig. 6).

a, b Representative U-ExM images of control and PfNuf2-cKO/KD (conditional knockout/knockdown) parasites labelled with the general protein dye NHS-PBS (a inverse greyscale) or NHS-BSA (b green), at early (30 hpi) and mid (36 hpi) schizont stages. The microtubules were labelled with anti-β-tubulin (yellow arrowheads, anti-β-tub, red), chromatin is marked by DAPI (blue), and the PfNuf2-HA is labelled with anti-HA (a green or b greyscale; evident only in control samples). Aqua arrowheads indicate the NHS-PBS-labelled outer centriolar plaque structures. The experiment was performed three times. c U-ExM images of PfNuf2-HA/GFP-PfCENH3 control and cKO/KD in early (30 hpi), middle (36 hpi), and late (38 hpi) stage schizonts labelled with NHS-PBS (inverse greyscale). The kinetochore protein, PfNuf2, is labelled with anti-HA (red) and the centromere protein, PfCENH3, is labelled with anti-GFP (green, GFP- PfCENH3). The anti-GFP signal overlaps with the anti-HA signal (white arrowheads) in control cells. Both signals are lost in cKO/KD parasites. The experiments were performed three times. Early (30 hpi), mid (36 hpi) and segmented schizonts (41 hpi) were labelled with NHS-PBS (d) or NHS-BSA (e). The outer centriolar plaque is labelled with anti-centrin (red, orange arrowheads). Both spindle microtubules (d green, white arrowheads) and subpellicular microtubules (e greyscale, aqua arrowheads) are labelled with anti-α-tubulin (anti-α-tub). The experiment was repeated three times. f Control and PfNuf2-disrupted segmented schizonts are labelled with NHS-PBS (inverse greyscale), highlighting the protein-dense rhoptries (purple arrowheads). The subpellicular microtubules are labelled with anti-β-tubulin (red), the chromatin with DAPI (blue) and the inner membrane complex with anti-PfGAP45 (anti-GAP45, green, magenta arrowheads). An NHS-PBS-labelled region in control cells likely represents the spindle remnant/ inner centriolar plaque (yellow arrowheads). In PfNuf2-cKO/KO parasites, a green arrowhead indicates an unusual NHS-PBS-labelled area inside the nucleus. The experiment was performed two times. g Anti-polyE (green) is concentrated at the minus end of subpellicular microtubules (white arrowheads) close to the apically located rhoptries. The experiment was performed two times. All images are presented as z-projections. All scale bars are 5 μm, except the zoom images, which are 2 μm. Additional images of PfNuf2-cKO/KO parasites are presented in Supplementary Figs. 13, 14 and 15.

Diagram illustrating the apparent locations of different parasite structures during mitosis; and disruption of organisation upon loss of the kinetochore components. (a) Hemispindle. (b) Mitotic spindle. (c) Extended spindle. (d) Divided nuclei. (e) Segmented schizont. In kinetochore-disrupted parasites, the mitotic spindle is disorganised, the inner and outer centriolar plaque structures are separated and the nexus between the mitotic machinery and the apical complex is lost.
In PfNuf2-cKO/KD parasites undergoing early (Fig. 5d, second row, Supplementary Fig. 14a, cKO/KD), mid (Fig. 5d, fourth row, Supplementary Fig. 14c, cKO/KD) and late rounds of nuclear division (Supplementary Fig. 14b, d, cKO/KD) and in segmented schizonts (Fig. 5e, cKO/KD), the arrangement of the centrin-labelled puncta (orange arrowheads) is disorganised. In the mid nuclear division, multiple centrin-labelled puncta are observed associated with one nucleus (Supplementary Fig. 14c, cKO/KD, Zoom 1), while other centrin puncta aggregate in regions of the cell distal to the nuclei (cKO/KD, Zoom 2). In later nuclear division, the association of the apical complex, nascent sub-pellicular microtubules (aqua arrowheads) and rhoptries (purple arrowheads) is maintained but the outer centriolar plaque (labelled with anti-centrin, orange arrowheads) is disconnected from the nucleus (Supplementary Fig. 14b, d, cKO/KD).
The subpellicular microtubules are associated with rhoptries (purple arrowheads) and the PfGAP45-labelled IMC (Fig. 5f, magenta arrowheads). The subpellicular microtubules (Fig. 5g, aqua arrowheads) are polyglutamated (anti-polyE, white arrowheads) but are separated from the nuclei (Fig. 5g, cKO/KD). An NHS-PBS-labelled structure that likely represents the remnant spindle and inner centriolar plaque (kinetochores and associated components) is present at the nuclear periphery in the control cells (Fig. 5f, control, yellow arrowheads). Regions located away from the nuclear periphery with a similar staining profile can be observed in the PfNuf2-cKO/KD parasites but we are unable to confirm the protein composition (Fig. 5f, g, cKO/KD, green arrowhead). Additional data showing different stages of schizogony and individual zoom-in panels, with NHS-PBS labelling are presented in Supplementary Fig. 15a (PfGAP45 labelling) and 15b (polyE labelling). These data confirm that cKO/KD of PfNuf2 results in a loss of the connection between the intranuclear spindle and the apical organelles, leading to aberrant segregation of the centriolar plaque, sub-pellicular microtubules and IMC away from the nuclei (Diagram in Fig. 6).
Discussion
Early electron microscopy studies revealed that spindle microtubules are nucleated from a nuclear membrane-spanning structure, referred to as the centriolar plaque, and showed that the spindle is decorated with kinetochores12,41,42. Following several rounds of nuclear division, the secretory and invasion machineries of the nascent merozoites are assembled in an apical complex, just above the site of the centriolar plaque. As segmentation proceeds, an apical complex is assembled and a nucleus is captured by each forming merozoite43. However, the physical basis for the coordination of these events was unclear.
In this study, we used U-ExM to examine the location and function of the outer kinetochore proteins, PfNDC80 and PfNuf2, and their interactions with other components of the mitotic machinery. The initiating step in mitosis is the formation of the hemispindle consisting of about five bundles of microtubules, emanating from a nuclear lumen-facing protein-dense structure (NHS-PBS-labelled in U-ExM)17,20,23. In our study, we see PfNDC80 and PfNuf2 are concentrated at the base of the hemispindle. Interestingly the centromere marker, PfCENH3, is also concentrated in the same region indicating pre-assembly of the kinetochore/ centromere complex prior to chromatin duplication. This may facilitate the capture of the uncondensed chromosomes. This pre-assembly contrasts with mammalian kinetochores that assemble only during mitosis42. The co-incidence of PfNDC80, PfNuf2 and PfCENH3 with the NHS-PBS-labelled inner centriolar plaque, suggests that these kinetochore/ centromere components are likely major components of this structure. The lateral association of the kinetochores with the minus end of the hemispindle microtubules may stabilise microtubule bundles and help establish the spindle44.
Following duplication of the outer centriolar plaque, a short mitotic spindle is established and PfNDC80, PfNuf2 and PfCENH3 relocate to the mid-plane of the spindle, consistent with the role of segregating sister chromatids20,23,37. The NHS-PBS-labelled region also moves along the spindle. The two outer centriolar plaque structures then migrate away from each other around the nuclear periphery, and an extended spindle is observed37. It is important to note that this is not the equivalent of the mammalian anaphase spindle as the chromatids have already been separated. At this stage, the kinetochore and centromere markers retract back to the region below the centrin puncta, and the NHS-PBS-labelled inner centriolar plaque structure is again evident. Taken together these data suggest that the NHS-PBS-labelled region in the nuclear lumen comprises the kinetochores and associated machinery. Dynamic movements of the kinetochores may help establish the spindle microtubules and help to achieve fidelity of DNA separation. The apparent plasticity of kinetochore organisation is consistent with previous live cell imaging studies in P. berghei and P. falciparum, as well as in other eukaryotic cells17,20,23.
In agreement with previous reports37,43,45, we confirmed that in later stages of schizogony, the outer centriolar plaque is positioned toward the apical complex (including rhoptries, IMC, PPM), which are in turn, position towards the periphery of the parent cell. Previous data for P. falciparum37,43,45 and T. gondii46,47 suggest that endoplasmic reticulum extensions close to the centriolar plaque generate the membrane material for the apical organelles. Based on U-ExM images, a physical link from the centriolar plaque to the apical prominence, the rhoptries, the Golgi and the apicoplast, has been suggested37.
Previous electron microscopy studies examined fully formed merozoites and observed that the sub-pellicular microtubules arise from a tubulin-based apical annulus, called the apical polar ring11,48,49. Interestingly, our analysis of the earlier stage schizonts suggests that cytoplasmic microtubules are first initiated from the cytoplasm-facing part of the centriolar plaque, although we cannot rule out nucleation from a closely associated nascent apical polar ring. The suggestion that the outer centriolar plaque is the initial site of nucleation, is consistent with previous work on stage II gametocytes, which lack an apical polar ring, in which cytoplasmic microtubules appear to nucleate from the outer region of the nuclear membrane-embedded centriolar plaque17. It is possible that the formation of the cytoplasmic microtubules helps position and orient the nascent merozoite, similar to the role played by aster microtubules in vertebrate cells50.
Conditional knockout/knockdown of PfNDC80 and PfNuf2 reveals their important roles in P. falciparum mitosis. Consistent with recent PfNDC80 mislocalisation data29, we show that both proteins are essential for parasite proliferation in the asexual blood stage. Disruption of individual kinetochore components is expected to result in defective binding to the centromeres, and thus a failure to capture the chromosomes during the division process. In vertebrate cells, the presence of chromosomes that remain unattached to kinetochores is detected by the cell, activating signalling pathways that stall the onset of anaphase51. Interestingly, disruption of PfNDC80 and PfNuf2 does not appear to prevent chromatin duplication or karyokinesis, although there is evidence for uneven inheritance of chromatin in the divided nuclei. Our data confirm that Plasmodium mitosis lacks robust checkpoints once mitosis has started, and that karyokinesis and cytokinesis are independently regulated processes35,52,53.
In this study, we show that disruption of the kinetochore components leads to unregulated mitosis. In the early rounds of nuclear division, aberrant spindles are observed that are disorganised and disconnected from the nuclear membrane. Some of the newly formed nuclei exhibit multiple centrin puncta, while other nuclei have none, indicating uneven inheritance of the centriolar plaque, and dysregulation of nuclear division. Of particular interest, the signal for the centromere marker, PfCENH3, is lost upon knockdown of kinetochore components. This suggests that kinetochore-based attachment of the chromosomes to the spindle microtubules is needed for integrity of the centromere complex.
During the final round of division, aberrant segmented merozoites are formed. The centrin-labelled outer centriolar plaque structure is disengaged from the cytoplasmic side of the nuclear membrane. Nonetheless, it remains connected to the apical organelles, such as the IMC and the rhoptries, and the sub-pellicular microtubules appear to be assembled from the disengaged centriolar plaque and the associated apical polar ring. The loss of this physical connection results in failure of the nascent merozoites to capture a nucleus. Intriguingly, the PfNDC80 and PfNuf2 cKO/KD segmented schizonts are still capable of egress despite the incomplete assembly of the merozoites. The data further highlight the independent regulation of karyokinesis and cytokinesis in P. falciparum and provide functional evidence that the position of the outer centriolar plaque directs the location of apical complex assembly and merozoite segmentation.
The effect of NDC80 knockdown has been assessed previously in the gametocyte and mosquito stages of P. yoelii30. PyNDC80-knockdown did not affect the formation of spindle microtubules during exflagellation, of the P. yoelii male gametocyte but it did cause a defect in chromosome segregation, leading to a decreased number of activated male gametes30. The difference in phenotype may be due to stage- or species-specific effects.
The centriolar plaque mislocalisation phenotype that we observed is similar to, but more severe than, the phenotype observed upon conditional knock-down of Nuf2 in T. gondii22. TgNuf2 disruption decreased the interaction between the cytoplasmic part of the centrosome equivalent and the centrocone (spindle pole) during tachyzoite cell division. Moreover the daughter cell did not inherit chromatin and the centromeres remained assembled and associated with the parent nucleus22. Disruption of spindle and kinetochore-associated proteins (SKA), including TgSKA1, TgSKA2 and TgSKA3, decreased the efficiency of the alignment and segregation of the centromeres and kinetochores into daughter cells23, while knockdown of the SCFFBXO1 E3 ubiquitin ligase also results in aberrant formation of the apical complex and the IMC54. Different phenotypes may be due to species-specific differences, such as the structurally different centriolar plaque in Plasmodium compared to the centrocone in Toxoplasma.
Less dramatic phenotypes have also been reported previously for disruptions of P. falciparum mini-chromosome maintenance complex binding-protein (MCMBP) and schizont egress antigen-1 (SEA1, a putative centromere-associated protein). PfMCMBP-deficient parasites displayed hemispindle and spindle microtubules that were 30% longer, and showed more branches35. PfSEA1- and PfMCMBP-deficient schizonts exhibited 20% and 17.6% akaryotic merozoites, respectively52,55. By contrast, PfNDC80-deficient mitotic schizonts exhibited 58% akaryotic merozoites.
In conclusion, this work provides insights into the role of the centriolar plaque as an important nexus between the intranuclear spindle microtubules and the apical complex - a connection that enables correct positioning of the mitotic spindle as well as positioning the apical complex and building the apical-basal polarity (diagram in Fig. 6). We show that deletion of the outer kinetochore components, PfNDC80 and PfNuf2, results in disruption of the centromere marker and partial failure of the spindle microtubules to segregate the chromosomes. This, in turn, leads to disengagement of the outer centriolar plaque from the nuclear envelope and loss of the connection between the apical complex and the nucleus (diagram in Fig. 6). The breaking of this important nexus leads to aberrant mitosis, resulting in akaryotic merozoites that are incapable of invading new RBCs. Our findings are reminiscent of a study in human cells, in which disruption of the centromere complex was shown to impact mitotic spindle pole integrity, leading to release of the microtubule minus-ends from the centrosome27. Our findings underscore the evolutionary conservation of pathways involving centromeres and centrosomes and highlight the value of using P. falciparum as a model for exploring fundamental questions in cell biology. Targeting the kinetochore/ centrosome nexus may provide a strategy to fight this very important human disease.
Methods
Parasite culture and synchronisation
Asexual blood stage P. falciparum parasites were cultured in RPMI-HEPES containing 0.25% AlbuMAXII and 5% human serum at 37 °C56. The parental parasite line NF54 DiCre used for transfection was maintained in 2.5 ug/mL Blasticidin S31. Late-stage parasites were enriched from culture using Percoll purification (GE Health) or magnetic purification (Miltenyi Biotec). Sorbitol synchronisation was used to obtain ring stage parasites57.
Plasmid constructs and transfection
Transgenic parasite lines were generated in which PfNDC80 and PfNuf2 were C-terminally tagged with a 3×HA tag and incorporated flanking loxP sites and a glmS riboswitch in the 3′UTR of the gene allowing for conditional knockout (cKO) of the gene and knockdown (cKD) of the transcript. The repair constructs were created by cloning a synthetic DNA fragment (HR1-intron-recodonised coding sequence, Genewiz) consisting of a 5′ homology region, an artificial intron containing a loxP site and the recodonised 3′ end of the gene into the BglII/PstI sites of the pGLMS-HA-glmS plasmid58. The 5′ homology region of PfNDC80 comprises 198 bp upstream of the start site and the first 171 bp of the coding sequence. For PfNuf2, 322 bp of the 5′ untranslated region and 36 bp of the coding sequence were used. These positions were chosen based on suitable Cas9 guide locations. All sequences downstream of the guide were recognised in the synthesised fragments. A 3′ homologous targeting sequence from the 3′ end (HR2) of PfNDC80 or PfNuf2 was PCR amplified (Supplementary Table 1) and cloned into the EcoRI and KasI sites of the plasmid. Plasmid DNA (50 μg) was linearised by overnight digestion with BglI and BglII enzymes, ethanol precipitated and resuspended in 30 μL TE buffer ready for transfection. Custom guides were designed and ordered as an Alt-R CRISPR-Cas9 crRNA (IDT™, Supplementary Table 2). Transfections were performed using the complexed Cas9 recombinant protein, guide RNAs and the linearised repair template59.
A previously generated NF54 parasite line stably expressing a rapamycin inducible split Cre recombinase31 was used as the parental strain in this work. These transfectants were maintained with 2.5 nM WR and 2.5 μg/mL Blasticidin S. Integration of the plasmids into the endogenous loci and loxP excision were confirmed by PCR (Supplementary Figs. 1b, c and 4c, e) using the primers described in Supplementary Table 1. See Supplementary Fig. 1a for schematic and primer locations. Uncropped gels are provided in Supplementary Fig. 16a, b, f, h.
To investigate the location of PfCENH3, we co-transfected the PfNuf2 and PfNDC80-HA transfectants with a plasmid encoding a GFP-PfCENH3 chimeric protein. The ydodh gene was cut from the pNLS-FRB-mCherry plasmid60 using the BamHI and HindIII restriction sites and directionally cloned into the BamHI and HindIII sites of the pPfNDC80-GFP-PfCENH3 construct17, replacing the selectable markers. The GFP-PfCENH3 plasmid was transfected into the PfNDC80 and PfNuf2 parental strain and maintained on 0.9 μΜ DHODH inhibitor (DSM1)61.
P. falciparum growth assays
Tightly synchronised (2-h window) ring-stage parasites were plated in triplicate into 24-well plates (2 mL/well) at 0.2–0.5% parasitemia (5% haematocrit). Parasite-infected RBCs were treated with either 100 nM rapamycin (41-h duration) and 2.5 mM glucosamine (continuously) or with 0.04% DMSO (41-h duration). Parasite samples were taken at 36 h from the first cycle and then every 41 h for 4 cycles. Parasitemia levels were assessed by flow cytometry. Parasite-infected RBCs were diluted to 0.01% haematocrit in 1×PBS and labelled with SYTO™ 61 for 15 min and analysed by flow cytometry62. Flow cytometry was performed on a BD FACSCanto™ II equipped with a High Throughput Sampler (HTS) and data was analysed using FlowJo software (version 10.7.1_CL)62.
Western blotting
Tightly synchronised schizonts were harvested at relevant time points and lysed with chilled saponin (0.03% (w/v) in PBS buffer) and incubated on ice for 15 min. The lysate was centrifuged at 3300 × g for 10 min, and the supernatant was removed and discarded. The parasite pellets were resuspended and washed three times in pre-chilled PBS containing an EDTA-free protease inhibitor cocktail (cOmplete, Roche). The samples were prepared for electrophoresis by resuspending the parasite pellets (10 μL) in Bolt LDS sample buffer (4×) (Invitrogen) (25 μL), Bolt sample reducing agent (10×) (Invitrogen) (10 μL) and chilled PBS containing protease buffer (65 μL). The samples were heated at 95 °C for 10 min and loaded into 4–20% Mini-PROTEAN® TGX™ Precast protein gel (BIO-RAD) and separated by electrophoresis in 1× Tris/Glycine/SDS buffer (BIO-RAD). Proteins were transferred from gels onto nitrocellulose membranes using the Trans-Blot® Turbo™ transfer system. The membranes were blocked in Intercept® (PBS) Blocking Buffer (LICORbio) for an hour at room temperature. Both primary and secondary antibodies were diluted in the blocking buffer. Primary antibodies: mouse anti-HA (Millipore; 05-904; LOT: 3005257; 1:500), mouse anti-GFP (TakaRa Living Colors® Av Monoclonal Antibody, JL-8 (632381); 1:500) and rabbit anti-Pfaldolase (abcam, ab207494; 1:3,000)63. Secondary antibodies: goat anti-mouse IRDye® 800CW (LICORbio; 926-32210; 1:10,000), donkey anti-rabbit IRDye® 680LT (LICORbio; 925-68023; 1:10,000). The membranes were incubated in primary antibodies overnight at 4 °C and secondary antibodies for 1 h at room temperature. The membranes were washed three times with phosphate buffered saline with 0.05% TWEEN® 20 (PBST) after incubations with primary and secondary antibodies, respectively. The membranes were imaged on an Odyssey® CLx imaging system (LICROBio). Uncropped gels are provided in Supplementary Fig. 16c, d, e, g, i.
Fluorescence microscopy
For the live cell imaging, tightly synchronised mature schizonts (41 hpi) were collected. The parasites were incubated with Hoechst 33342 (5 μg/mL, Invitrogen) to label DNA, and Tubulin Tracker Deep Red (1 μM, Invitrogen) to label the microtubules, for 15 min at 37 °C, before imaging. The live cell data were acquired on a restorative wide field deconvolution microscope (DeltaVision DV Elite, Applied Precision) equipped with a 100× oil immersion objective (numerical aperture (NA) = 1.46). The images were acquired and deconvolved using the softWoRx 6.1 software (Applied Precision). The images were processed in FIJI software (version 2.3.0). All live cell images are displayed as maximum intensity projections (using FIJI) of full z-stacks.
For immunofluorescence microscopy, highly synchronised mitotic (36 hpi) schizonts were sampled onto coverslips pre-coated with 0.1 mg/mL erythroagglutinating phytohemagglutinin (PHAE) and incubated at 37 °C for 20 min. The cover slips were washed three times with PBS, prior to being fixed in 2% (v/v) formaldehyde in PBS for 30 min at 37 °C. After fixation, the coverslips were washed three times in PBS and bound cells were permeabilized with 0.2% (v/v) Triton-X 100 (in PBS) for 20 min at room temperature. The coverslips were again washed three times with PBS and incubated with the primary antibodies (diluted in 3% BSA/PBS) for 3 h at room temperature. The following primary antibodies were used: rabbit anti-HA (Sigma-Aldrich; H6908; 1:200) and mouse anti-β-tubulin (Sigma-Aldrich; T5201; clone TUB 2.1; 1:200). After washing three times with PBS, the coverslips were incubated for 1 h with secondary antibodies (diluted in 3% BSA/PBS). The following secondary antibodies were used: Alexa Fluor 568 goat anti-mouse IgG (H + L) (Invitrogen; A-11004; 1:500) and Alexa Fluor 488 goat anti-rabbit lgG (H + L) (Life technologies; A-11008; 1:500). The parasite DNA was labelled with DAPI solution (2 μg/mL in PBS, Thermo Scientific, 62248) for 15 min at room temperature and washed three times with PBS. The coverslips were mounted in anti-fade solution (90% glycerol/0.2% w/v p-phenylenediamine) on glass slides. The images were acquired and processed as described above. All immunofluorescence images are displayed as maximum intensity projections (using FIJI software) of full z-stacks.
Ultrastructure expansion microscopy
U-ExM was performed following published protocols33,34,35,36 with some adaptions as follows. Coverslips (22 × 22 mm # 1.5 mm; Lecia, 3800106) were cut into four square pieces (11 × 11 mm). The coverslips were incubated with poly-D-lysine (Gibco, A38904-01, 0.1 mg/mL) for 1 h at 37 °C, washed three times with MilliQ water, dried under nitrogen gas and placed in a 24-well plate. Tightly synchronised mitotic (30, 34, 36, 38 hpi) or mature segmented schizonts (41 hpi; with a 2-h window) were harvested by Percoll purification, resuspended in PBS and overlayed onto the precoated coverslip and incubated for 15 min at 37 °C. The unbounded cells were carefully removed from the well and the bound cells fixed with 4% (v/v) formaldehyde in PBS for 15 min at 37 °C. After fixation, the coverslips were washed three times with prewarmed PBS and incubated in 1.4% (v/v) formaldehyde/2% (v/v) acrylamide in PBS overnight at 37 °C.
A monomer solution comprising sodium acrylate (19% w/w; Sigma-Aldrich, 408220), acrylamide (10% v/v; Sigma-Aldrich, A4058) and N, N’-methylenebisacrylamide (BIS; 2% v/v; Sigma-Aldrich, M1533) in PBS was prepared 24 h before use and stored at −20 °C. For parasite embedding in the gel, 5 μL of pre-chilled 10% (v/v) tetramethylenediamine (TEMED) (Thermo Scientific, 17919) and 5 μL of 10% (w/v) ammonium persulfate (APS) (VWR Chemicals BDH, BDH9216) were added to 90 μL of monomer solution and vortexed for 3–5 s. Parafilm squares larger than the coverslip were placed on ice and are used as a base for gel setting and sample embedding. Thirty-five microliters of the gel mixture were added onto the prechilled Parafilm and the coverslip (cell side face down) placed on top of the gel mixture and incubated at 37 °C for 1 h. The gel and coverslip were removed from the mould and incubated in denaturation buffer (200 mM sodium dodecyl sulphate (SDS), 200 mM NaCl, 50 mM Tris, pH = 9) in a 6-well plate and incubated, shaking for 15 min at room temperature. The gels were detached from the coverslips and transferred into 1.5 mL Eppendorf tubes containing the denaturation buffer and incubated at 95 °C for 90 min. The denatured gels were transferred into a clean dish (10 cm) containing MilliQ water for the first round of expansion. The dishes were placed on the shaker for 30 min at room temperature, the MilliQ water exchanged, and the dishes again placed on the shaker. This step was performed three times in total. After expansion, the gels were re-equilibrated by incubating with PBS for 15 min and then blocked in 3% BSA/PBS for 30 min at room temperature. After blocking, the gels were incubated with primary antibodies at room temperature overnight on the shaker (Supplementary Table 3). Following antibody incubation, the gels were washed three times with PBST (10 min/wash), prior to addition of the secondary antibodies (Supplementary Table 3), general protein dye DyLight™ 488 NHS Ester (Thermo Fisher, 46402) and DAPI for 2.5 h incubation in the dark with gentle shaking at room temperature. For experiments where membrane staining was required, the secondary antibodies, DAPI (200 μg/mL final concentration) and NHS Ester dye (10 μg/mL final concentration) were diluted in 3% BSA/PBS solution, while for more generalised protein labelling, the secondary antibodies, DAPI (200 μg/mL final concentration) and NHS Ester dye (10 μg/mL final concentration) were diluted in the PBS. Following secondary antibody incubation, the gels were washed three times with PBST (10 min/wash) and transferred to a Petri dish containing MilliQ water and incubated 30 min. The MilliQ water was exchanged three times.
To mount and image the expanded gels, 35 mm coverslip dishes (uncoated, γ-Irradiated, MarTek Corporation, P35G-2-14-C-GRID, or uncoated, #1.5 polymer coverslip, hydrophobic, sterilised ibidi, 81151) were coated with 0.5 mL poly-D-lysine for 1 h at 37 °C, washed with MilliQ water and dried under nitrogen gas. The gels were transferred to precoated glass bottom dishes, with the cell side down. The expansion of each gel was calculated by measuring the length and width of gel prior to and after expansion.
U-ExM data were acquired using a Zeiss Elyra LSM880/ Axio Observer Z1 LSM980 microscope equipped with an Airyscan detector (Carl Zeiss) in “SR” mode and using a 100 × oil immersion objective (numerical aperture = 1.4) or 63 × oil immersion objective (numerical aperture = 1.4) or a Zeiss Axio Observer LSM900 with an Airyscan 2 detector with a 63x APOCHROMAT objective (numerical aperture = 1.4). The images were acquired as Z-stacks (0.2 μm or 0.3 μm per slice) and processed using the Airyscan Processing tool within the Zen black software (version 2.3) or Zen blue software (version 3.8). All U-ExM images are displayed as maximum intensity projections (using FIJI software) of multiple z-slices (thickness ranges from 2 μm to 3 μm). Nuclear volumes (expanded) were analysed in segmented schizonts, using the Imaris software (version 10.0.0). Airyscan processed images were converted to Imaris file format (.ims) using Imaris File Converter (version 10.0.1). Prior to 3D reconstruction of the nuclei, the membrane channel (NHS-BSA staining) was subtracted from the nucleus channel (DAPI staining) using the Channel Arithmetics tool to improve the boundary detection between the nuclei. The remaining nucleus signal (i.e., DAPI minus NHS-BSA) was used to reconstruct a 3D object using the ‘surface creation wizard’. In all images, an automatic threshold was used to avoid user bias in volume measurement and the reconstructed 3D objects were inspected manually to avoid over/under detection in counting.
Statistical analysis
All data are presented as means plus and minus the standard deviation (SD), with specific details outlined in the Fig. legends. Each live-cell fluorescence experiment and flow cytometry growth assay were performed at least three times. Each PCR and Western blot were performed at least two times. The data displayed are representative of these experiments. Not all expansion microscopy experiments were repeated three times, due to the large volumes of antibodies required and availability. This is recorded in the Fig. legends. The cell numbers analysed in each experiment are presented in the Fig. legends. The analysis of the volumes of the (expanded) nuclei is described in the Fig. legend. Statistical significance was analysed using Welch’s t-test with 95% confidence intervals as indicated in the Fig. legends. Statistical analyses were conducted using GraphPad Prism 8.
Transmission electron microscopy
Segmented schizonts (41 hpi) were enriched by Percoll purification. The enriched parasite-infected RBCs were fixed in 1 mL of 2% (v/v) glutaraldehyde and 2% (v/v) formaldehyde in 1 × PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2) and incubated overnight at 4 °C. The cells were washed for 5 min in 1.5 × PHEM buffer, centrifuged at 500 × g, the buffer was replaced, and the infected RBCs were washed a further 2 times. The parasites were post-fixed in 1% (w/v) osmium tetroxide in 1 × PHEM buffer for 30 min in the dark. The infected RBC pellet was washed with 1.5 × PHEM buffer three times as described above. The infected RBC pellet was next incubated in 1% (w/v) tannic acid in 1 × PHEM buffer for 20 min. Samples were washed three times with MilliQ water, with centrifugation as described above. The infected RBCs were next incubated in the dark in 1% (w/v) aqueous uranyl acetate for 20 min, before 3 rounds of washing in MilliQ. Following washing, samples were dehydrated in an ascending ethanol series (30%, 50%, 70%, 90%, 100%, 100%) (5 min each condition). Samples were next incubated in two changes of absolute acetone for 10 min each. The cells were gradually infiltrated with acetone: medium grade Procure 812 resin (2:1, 1:2) for 2 h, followed by pure resin incubation overnight and a final change of the pure resin the next day for another 3 h, prior to polymerisation for 48 h at 60 °C.
After polymerisation, samples were trimmed and 70 nm sections generated using an ultramicrotome (Lecia EM UC7; Leica Microsystems, Wetzlar, Germany). The sections were collected on formavar-carbon 100-mesh and imaged on a FEI Tecnai F30 electron microscope (FEI company, Hillsboro, OR) at an accelerating voltage of 200 keV.
Array tomography
For array tomography, 120 nm serial sections were generated and collected on a hydrophilized silicon wafer. Samples were post-stained with 2% aqueous uranyl acetate for 10 min and Reynold’s lead citrate for 10 min. Backscattered field emission scanning electron microscopy was performed using a Zeiss Sigma operating at 3 keV accelerating voltage and 5 mm working distance. Images were normalised and binned in FIJI and automatically aligned using the StackReg plugin64. Features of interest were segmented, visualised and analysed using IMOD (version 4.9.9)65.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Additional data are available in Supplementary Information. Source data are provided. Source data are provided with this paper.
References
World Malaria Report. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO (2022).
Conrad, M. D. et al. Evolution of partial resistance to artemisinins in malaria parasites in Uganda. N. Engl. J. Med. 389, 722–732 (2023).
Rogerson, S. J. et al. Identifying and combating the impacts of COVID-19 on malaria. BMC Med. 18, 239 (2020).
Beaudoin, R. L. & Aikawa, M. Primaquine-induced changes in morphology of exoerythrocytic stages of malaria. Science 160, 1233–1234 (1968).
Gerald, N., Mahajan, B. & Kumar, S. Mitosis in the human malaria parasite Plasmodium falciparum. Eukaryot. Cell 10, 474–482 (2011).
McDonald, J. & Merrick, C. J. DNA replication dynamics during erythrocytic schizogony in the malaria parasites Plasmodium falciparum and Plasmodium knowlesi. PLoS Pathog. 18, e1010595 (2022).
Azimzadeh, J. & Bornens, M. Structure and duplication of the centrosome. J. Cell Sci. 120, 2139–2142 (2007).
Pines, J. & Rieder, C. L. Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3, E3–E6 (2001).
Arnot, D. E., Ronander, E. & Bengtsson, D. C. The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int. J. Parasitol. 41, 71–80 (2011).
Moore, J. & Sinden, R. E. Fine structure of Plasmodium mexicanum. J. Parasitol. 60, 825–834 (1974).
Bannister, L. H., Hopkins, J. M., Fowler, R. E., Krishna, S. & Mitchell, G. H. A brief illustrated guide to the ultrastructure of Plasmodium falciparum asexual blood stages. Parasitol. Today 16, 427–433 (2000).
Prensier, G. & Slomianny, C. The karyotype of Plasmodium falciparum determined by ultrastructural serial sectioning and 3D reconstruction. J. Parasitol. 72, 731–736 (1986).
Baron, A. T., Greenwood, T. M. & Salisbury, J. L. Localization of the centrin-related 165,000-Mr protein of PtK2 cells during the cell cycle. Cell Motil. Cytoskeleton 18, 1–14 (1991).
Mahajan, B. et al. Centrins, cell cycle regulation proteins in human malaria parasite Plasmodium falciparum. J. Biol. Chem. 283, 31871–31883 (2008).
Roques, M. et al. Plasmodium centrin PbCEN-4 localizes to the putative MTOC and is dispensable for malaria parasite proliferation. Biol. Open 8, bio036822 (2019).
Simon, C. S. et al. An extended DNA-free intranuclear compartment organizes centrosome microtubules in malaria parasites. Life Sci. Alliance 4, e202101199 (2021).
Li, J. et al. Repurposing the mitotic machinery to drive cellular elongation and chromatin reorganisation in Plasmodium falciparum gametocytes. Nat. Commun. 13, 5054 (2022).
Wenz, C. et al. An Sfi1-like centrin-interacting centriolar plaque protein affects nuclear microtubule homeostasis. PLoS Pathog. 19, e1011325 (2023).
Reininger, L., Wilkes, J. M., Bourgade, H., Miranda-Saavedra, D. & Doerig, C. An essential Aurora-related kinase transiently associates with spindle pole bodies during Plasmodium falciparum erythrocytic schizogony. Mol. Microbiol. 79, 205–221 (2011).
Zeeshan, M. et al. Real-time dynamics of Plasmodium NDC80 reveals unusual modes of chromosome segregation during parasite proliferation. J. Cell Sci. 134, jcs245753 (2021).
Wei, R. R., Sorger, P. K. & Harrison, S. C. Molecular organization of the Ndc80 complex, an essential kinetochore component. Proc. Natl. Acad. Sci. USA 102, 5363–5367 (2005).
Farrell, M. & Gubbels, M. J. The Toxoplasma gondii kinetochore is required for centrosome association with the centrocone (spindle pole). Cell Microbiol. 16, 78–94 (2014).
Brusini, L., Dos Santos Pacheco, N., Tromer, E. C., Soldati-Favre, D. & Brochet, M. Composition and organization of kinetochores show plasticity in apicomplexan chromosome segregation. J. Cell Biol. 221, e202111084 (2022).
Alushin, G. M. et al. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467, 805–810 (2010).
Sundin, L. J., Guimaraes, G. J. & DeLuca, J. G. The NDC80 complex proteins Nuf2 and Hec1 make distinct contributions to kinetochore–microtubule attachment in mitosis. Mol. Biol. Cell 22, 759–768 (2011).
Hoeijmakers, W. A. et al. Plasmodium falciparum centromeres display a unique epigenetic makeup and cluster prior to and during schizogony. Cell Microbiol. 14, 1391–1401 (2012).
Gemble, S. et al. Centromere dysfunction compromises mitotic spindle pole integrity. Curr. Biol. 29, 3072–3080 e3075 (2019).
Zeeshan, M. et al. Plasmodium SAS4: basal body component of male cell which is dispensable for parasite transmission. Life Sci. Alliance 5, e202101329 (2022).
Kimmel, J. et al. Gene-by-gene screen of the unknown proteins encoded on Plasmodium falciparum chromosome 3. Cell Syst. 14, 9–23 e27 (2023).
Yang, S. et al. EB1 decoration of microtubule lattice facilitates spindle-kinetochore lateral attachment in Plasmodium male gametogenesis. Nat. Commun. 14, 2864 (2023).
Wilde, M. L. et al. Protein Kinase A is essential for invasion of Plasmodium falciparum into human erythrocytes. mBio 10, e01972–19 (2019).
Prommana, P. et al. Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PloS One 8, e73783 (2013).
Bertiaux, E. et al. Expansion microscopy provides new insights into the cytoskeleton of malaria parasites including the conservation of a conoid. PLoS Biol. 19, e3001020 (2021).
Gambarotto, D., Hamel, V. & Guichard, P. Ultrastructure expansion microscopy (U-ExM). Methods Cell Biol. 161, 57–81 (2021).
Liffner, B. & Absalon, S. Expansion microscopy reveals Plasmodium falciparum blood-stage parasites undergo anaphase with a chromatin bridge in the absence of mini-chromosome maintenance complex binding protein. Microorganisms 9, 2306 (2021).
Rashpa, R. & Brochet, M. Expansion microscopy of Plasmodium gametocytes reveals the molecular architecture of a bipartite microtubule organisation centre coordinating mitosis with axoneme assembly. PLoS Pathog. 18, e1010223 (2022).
Liffner, B. et al. Atlas of Plasmodium Falciparum intraerythrocytic development using expansion microscopy. eLife 12, RP88088 (2023).
Liffner, B. & Absalon, S. Hand-in-hand advances in microscopy and Plasmodium nuclear biology. Trends Parasitol. 38, 421–423 (2022).
Read, M., Sherwin, T., Holloway, S. P., Gull, K. & Hyde, J. E. Microtubular organization visualized by immunofluorescence microscopy during erythrocytic schizogony in Plasmodium falciparum and investigation of post-translational modifications of parasite tubulin. Parasitology 106, 223–232 (1993).
Baum, J. et al. A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J. Biol. Chem. 281, 5197–5208 (2006).
Aikawa, M. & Beaudoin, R. L. Studies on nuclear division of a malarial parasite under pyrimethamine treatment. J. Cell Biol. 39, 749–754 (1968).
Cheeseman, I. M. & Desai, A. Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46 (2008).
Bannister, L. H., Hopkins, J. M., Fowler, R. E., Krishna, S. & Mitchell, G. H. Ultrastructure of rhoptry development in Plasmodium falciparum erythrocytic schizonts. Parasitology 121, 273–287 (2000).
Matković, J. et al. Kinetochore- and chromosome-driven transition of microtubules into bundles promotes spindle assembly. Nat. Commun. 13, 7307 (2022).
Rudlaff, R. M., Kraemer, S., Marshman, J. & Dvorin, J. D. Three-dimensional ultrastructure of Plasmodium falciparum throughout cytokinesis. PLoS Pathog. 16, e1008587 (2020).
Francia, M. E. & Striepen, B. Cell division in apicomplexan parasites. Nat. Rev. Microbiol. 12, 125–136 (2014).
Harding, C. R. & Frischknecht, F. The riveting cellular structures of Apicomplexan parasites. Trends Parasitol. 36, 979–991 (2020).
Fowler, R. E., Fookes, R. E., Lavin, F., Bannister, L. H. & Mitchell, G. H. Microtubules in Plasmodium falciparum merozoites and their importance for invasion of erythrocytes. Parasitology 117, 425–433 (1998).
Hanssen, E. et al. Electron tomography of Plasmodium falciparum merozoites reveals core cellular events that underpin erythrocyte invasion. Cell Microbiol 15, 1457–1472 (2013).
Tang, N. & Marshall, W. F. Centrosome positioning in vertebrate development. J. Cell Sci. 125, 4951–4961 (2012).
Cleveland, D. W., Mao, Y. & Sullivan, K. F. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421 (2003).
Absalon, S. & Dvorin, J. D. Depletion of the mini-chromosome maintenance complex binding protein allows the progression of cytokinesis despite abnormal karyokinesis during the asexual development of Plasmodium falciparum. Cell Microbiol. 23, e13284 (2021).
Ganter, M. et al. Plasmodium falciparum CRK4 directs continuous rounds of DNA replication during schizogony. Nat. Microbiol. 2, 17017 (2017).
Rashpa, R., Klages, N., Schvartz, D., Pasquarello, C. & Brochet, M. The Skp1-Cullin1-FBXO1 complex is a pleiotropic regulator required for the formation of gametes and motile forms in Plasmodium berghei. Nat. Commun. 14, 1312 (2023).
Perrin, A. J. et al. Malaria parasite schizont egress antigen-1 plays an essential role in nuclear segregation during schizogony. mBio. 12, 10–1128 (2021).
Foley, M., Deady, L. W., Ng, K., Cowman, A. F. & Tilley, L. Photoaffinity labeling of chloroquine-binding proteins in Plasmodium falciparum. J. Biol. Chem. 269, 6955–6961 (1994).
Lambros, C. & Vanderberg, J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 (1979).
Dumont, L. et al. The metabolite repair enzyme phosphoglycolate phosphatase regulates central carbon metabolism and fosmidomycin sensitivity in Plasmodium falciparum. mBio. 10, 10–1128 (2019).
Collier, S. et al. Plasmodium falciparum formins are essential for invasion and sexual stage development. Commun. Biol. 6, 861 (2023).
Birnbaum, J. et al. A genetic system to study Plasmodium falciparum protein function. Nat. Methods 14, 450–456 (2017).
Phillips, M. A. et al. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 51, 3649–3653 (2008).
Klonis, N. et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. USA 108, 11405–11410 (2011).
Hollin, T. et al. Functional genomics of RAP proteins and their role in mitoribosome regulation in Plasmodium falciparum. Nat. Commun. 13, 1275 (2022).
Thévenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Acknowledgements
We thank Professor Rita Tewari, the University of Nottingham, UK and Professor Jing Yuan, Xiamen University, China, for advice. We acknowledge the facilities at the Biological Optical Microscopy Platform and the Flow Cytometry Platform, The University of Melbourne, the Microscopy Resources on the North Quad (MicRoN) core at Harvard Medical School, and the University of Sydney Microscopy & Microanalysis facilities of Microscopy Australia. For research support, we thank the Australian Research Council (FL150100106 to L.T.), the National Health and Medical Research Council of Australia (APP1098992 to L.T.) and the NIH (5R01AI167570 to M.T.D). B.L. is supported by an American Heart Association Postdoctoral Fellowship (23POST1011626).
Author information
Authors and Affiliations
Contributions
Conceptualisation: J.L., G.J.S., B.L, S.A., M.W.A.D., and L.T.; Investigation: J.L., G.J.S., and E.C.; Analysis: J.L., G.J.S., F.B., E.C., M.W.A.D., and L.T.; Funding acquisition: F.B. and L.T.; Writing: J.L., G.J.S., F.B., E.C., B.L., M.T.D., S.A., M.W.A.D., and L.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Shami, G.J., Liffner, B. et al. Disruption of Plasmodium falciparum kinetochore proteins destabilises the nexus between the centrosome equivalent and the mitotic apparatus. Nat Commun 15, 5794 (2024). https://doi.org/10.1038/s41467-024-50167-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50167-6
- Springer Nature Limited