
Abstract
Prolific breeds of domestic sheep (Ovis aries) are important genetic resources due to their reproductive performance, which is characterized by multiple lambs per birth and out-of-season breeding. However, the lack of a comprehensive understanding of the genetic mechanisms underlying the important reproductive traits, particularly from the evolutionary genomics perspective, has impeded the efficient advancement of sheep breeding. Here, for the first time, by performing RNA-sequencing we built a de novo transcriptome assembly of ovarian and endometrial tissues in European mouflon (Ovis musimon) and performed an mRNA–miRNA integrated expression profiling analysis of the wild species and a highly prolific domestic sheep breed, the Finnsheep. We identified several novel genes with differentially expressed mRNAs (e.g., EREG, INHBA, SPP1, AMH, TDRD5, and ZP2) between the wild and domestic sheep, which are functionally involved in oocyte and follicle development and fertilization, and are significantly (adjusted P-value < 0.05) enriched in the Gene Ontology (GO) terms of various reproductive process, including the regulation of fertilization, oogenesis, ovarian follicle development, and sperm–egg recognition. Additionally, we characterized 58 differentially expressed miRNAs and 210 associated target genes that are essential for the regulation of female reproduction cycles through specific regulatory networks [e.g., (miR-136, miR-374a, miR-9-5p)-(EREG, INHBA)]. Furthermore, our integrated mRNA and miRNA expression profiling analysis elucidated novel direct and indirect miRNA/mRNA causal regulatory relationships related to the reproductive traits of the Ovis species. This study provides in-depth insights into the genomic evolution underlying the reproductive traits of the Ovis species and valuable resources for ovine genomics.
Similar content being viewed by others


Introduction
Reproduction is an important but complex biological process in mammals. From the physiological perspective, the reproductive performance of a female is attributable to a series of biological events occurring between oocyte maturation and birth (Vinet et al. 2012). An increased number of offspring is largely associated with increased ovulation rates during the estrous cycle and subsequent successful embryonic and fetal development (Vinet et al. 2012; Warriach et al. 2015). The ovary and uterus are two essential parts of the female reproductive tract. Function of the ovary includes the production of oocytes, excretion of reproductive hormones, and maintenance of female estrus cycles (Peters and McNatty 1980). The endometrium, which is the inner-most epithelial layer of the mammalian uterus, functions to prevent adhesions between the opposing walls of the myometrium, thereby maintaining the patency of the uterine cavity (Fazleabas and Strakova 2002). Therefore, the ovary, endometrium, and their communications can directly affect female reproductive success (Peters and McNatty 1980; Fazleabas and Strakova 2002; Wira et al. 2015). From the evolutionary perspective, the reproductive success of females is optimally balanced by the frequency of the estrous cycle per year, time between parturition and first ovulation, number and size of offspring, and time at which sexual maturity is reached (Walker et al. 2008; Bronson 2009; Barrett et al. 2009).
During and after domestication, livestock reproductive traits have been continuously selected. Wild sheep such as Asian mouflon Ovis orientalis and European mouflon Ovis musimon typically show pronounced seasonal estrous in all reproductive parameters and are typically monotonous (Lincoln 1989; Santiago-Morenoa et al. 2000; Garel et al. 2005). After domestication, many sheep (Ovis aries) breeds continue to maintain seasonal estrus and have one lamb per litter, while several prolific breeds typically demonstrate the capability of non-seasonal breeding and have multiple lambs per litter, which could be ascribed to long-term artificial and natural selection (Rasali et al. 2006). Additionally, prolific breeds of domestic sheep show much earlier sexual maturation (as early as 6 months age) than wild sheep (more than 1.5 years age; Cugnasse et al. 1985; Garel et al. 2005). Early genetic mapping studies have detected a few quantitative trait loci (QTLs) and functional genes (e.g., BMPR1B, BMP15, and GDF9) associated with high prolificacy in various sheep breeds (Hanrahan et al. 2004; Bodin et al. 2007; Abdoli et al. 2013). However, to our knowledge no study has explored the genetic mechanisms underlying the development of reproductive traits from wild to domestic sheep.
Transcriptome profiling using next-generation sequencing has been successfully used to measure the levels of transcripts and their isoforms (Wang et al. 2009). In livestock, mRNA profiling has been used to characterize large-scale gene expression patterns and identify differentially expressed genes that are specifically associated with production traits (Mortazavi et al. 2008; Jiang et al. 2014; Suravajhala et al. 2016; Clark et al. 2017). Additionally, microRNA (miRNA) profiling has successfully used to identify particular miRNAs, predict their target genes, and analyze their potential biological functions (Ambros 2004; Bartel 2009; McDaneld 2009; Liu et al. 2014). In sheep, previous RNA profiling investigations have revealed novel mRNAs, miRNAs, relevant functional genes, and variants associated with a variety of traits, including high prolificacy traits such as multiple births and non-seasonal breeding (Di et al. 2014; Pokharel et al. 2015; Hu et al. 2016). However, integrated analyses of large-scale mRNA and miRNA expression profiling in two (e.g., ovary and endometrium) or more female reproductive tissues from sheep are scarce (but see Miao and Qin 2015; Pokharel et al. 2018). Additionally, to date no full-length transcriptome has been available for any wild sheep, which has impeded studies investigating the genomic evolution and functional diversification of specific miRNA genes (Liu et al. 2008; Berezikov 2011; Ameres and Zamore 2013) between wild and domestic sheep.
In this study, we performed genome-wide profiling of the mRNA and miRNA in ovarian and endometrial tissues in European mouflon and a prolific breed of domestic sheep, the Finnsheep (Fig. S1 and Table S1). In particular, for the first time we reconstructed a de novo transcriptome of wild sheep (Fig. S2 and Table S1) using the Trinity platform, which provides a novel algorithm for assembling full-length transcripts (Grabherr et al. 2011; Haas et al. 2013). The European mouflon is considered one of the close wild relatives to domestic sheep (Hiendleder et al. 2002; Lv et al. 2015). The Finnsheep, which is a native sheep breed from Finland, is well-known worldwide due to its excellent prolific performance, including its early maturity (at c. 6 months of age), multiple lamb births (typically 3–5 lambs per birth, and the recorded highest number of lambs is 9), and non-seasonal breeding (Maijala 1996; Li et al. 2011; Mullen and Hanrahan 2014). Nevertheless, early mRNA and miRNA studies investigating the prolificacy traits in Finnsheep have included very limited sample sizes, and the analyses of mRNAs and miRNAs were performed separately (Pokharel et al. 2015; Hu et al. 2016). By performing integrated and comparative analyses of the data within and between species, our aims are threefold: (i) to reveal the genetic mechanisms underlying the evolution of the reproduction traits in species of the genus Ovis; (ii) to decipher the genetic architecture of the high prolificacy traits in a domestic sheep breed; and (iii) to create novel mRNA and miRNA genomic resources associated with reproductive traits in wild and domestic sheep species. Our results provide a comprehensive understanding of the genetic regulatory network responsible for the high prolificacy traits and molecular mechanisms underlying their evolution in Ovis species, which could also be applied to other mammals.
Materials and methods
Sample collection, RNA isolation, and sequencing
All animal handling was carried out under the license approved by the Southern Finland Animal Experiment Committee (approval No. ESAVI/5027/04.10.03/2012). From the Finnsheep, six ovarian samples (F1-O, F2-O, F3-O, F4-O, F5-O, and F6-O) were collected from six ewes (F1, F2, F3, F4, F5, and F6), and two endometrial samples (F2-E and F3-E) were taken from F2 and F3 individuals. The ewes in the experiment were mature, with an approximate age and weight of 4.25 years and 71.2 kg, respectively. The ovarian samples were collected at the follicular growth phase based on hormonal profiles, and endometrial samples were collected during early pregnant stage after ovulation (Pokharel et al. 2018). Three European mouflon ewes (M1, M2, and M3) were killed by hunters in autumn of 2013 (M1 and M2) and 2014 (M3). To obtain comparable sample sizes between the wild and domestic sheep, we collected six ovarian samples (M1-OA, M1-OB, M2-OA, M2-OB, M3-OA, and M3-OB) representing two replicates (coded as -OA and -OB) from each of the three European mouflons. Two endometrial samples (M2-EA and M2-EB) were taken by cytobrush from both uterine horns of the older individual M2 (5–6 years old), and we did not take endometrial samples from the other two younger wild sheep. We did not have any information about the stage of the cycle in the mouflons except that the older one (M2) had a corpus luteum. In total, 16 samples consisting of 12 ovarian samples and 4 endometrial samples were included in the mRNA profiling analysis. Additionally, all 16 samples were included in the miRNA profiling analysis, except for M2-EA (Table S1).
After the sample collection, the tissue samples of domestic sheep were quickly stored in RNAlater reagent (Ambion/Qiagen, Valencia, CA, USA) per the manufacturer’s instructions and transported to the laboratory. For the European mouflons, the collected samples were stored in RNAlater within 1 h of death due to the forest areas. The tissue samples were then transferred to be stored at −80°C in laboratory until extraction. We used an RNeasy plus mini kit (Qiagen, Valencia, CA, USA) to extract the mRNA and miRNA from the tissues according to the manufacturer’s protocol. The RNA concentration and RNA integrity number were measured using a Bioanalyzer 2100 (Agilent Technologies, Waldbronn, Germany). Libraries of mRNAs and miRNAs were prepared using the Illumina’s TruSeq library preparation kits and sequenced using Illumina Hiseq2000 at the Finnish Microarray and Sequencing Center, Turku, Finland. The mRNA libraries were sequenced using 100 base-pair (bp) paired-end sequencing chemistry, whereas the miRNAs were sequenced using the single-end 50 bp approach.
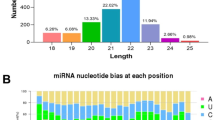
Quality control and mapping
FastQC v.0.11.4 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to examine the quality of the raw RNA-Seq (both the mRNA-Seq and miRNA-Seq) reads, and the adapter sequences were removed using the program Cutadapt v.1.9.1 (Martin 2011). The obtained trimmed mRNA-Seq reads were then mapped against the sheep reference genome (Oar v.4.0) supported by the gene annotation file of Ensembl release 83 using the program TopHat v.2.0.8b (Trapnell et al. 2012). For the miRNA-Seq data, the program PRINSEQ-LITE v.0.20.4 (Schmieder and Edwards 2011) was used for further miRNA read preprocessing (including filtering for reads containing ambiguous reads, data reformatting, and adapter trimming). Clean reads between 18 and 26 nucleotides (nt) in length were used for the subsequent analysis. Then, the Bowtie-build tool was used to build an index for the genome alignment using the sheep genome assembly Oar v.3.1 (ftp://ftp.ensembl.org/pub/release-83/fasta/ovis_aries/dna/), and the clean reads (18–26 nt) were mapped to the index using the program Bowtie v.2.2.1 (Langmead et al. 2009). The bioinformatics pipeline used to analyze the mRNA-Seq and miRNA-Seq data is summarized in Fig. S1.
De novo transcriptome assembly and annotation of European mouflon
We used the Trinity v.2.1.1 package (Grabherr et al. 2011) to construct a de novo transcriptome assembly of European mouflon using the ovarian (M1-OA, M1-OB, M2-OA, M2-OB, M3-OA, and M3-OB) and endometrial (M2-EA and M2-EB) mRNA sequences. The quality of the de novo assembly was assessed by examining the proportion of reads mapped to the assembly. We used the RSEM (RNA-Seq by expectation-maximization) tool (Li and Dewey 2011) to estimate the number of RNA-Seq fragments that map to each contig in the individual M2 (M2-OA, M2-OB, M2-EA, and M2-EB), and detected the differentially expressed genes between the two types of tissues, i.e., ovary and endometrium, using the EdgeR package (Robinson et al. 2010). Unigenes that had |log2(fold change)| ≥ 2 and a false discovery rate ≤ 0.01 were considered significantly differentially expressed. The differentially expressed unigenes were mapped to the NCBI nt collection database using the BlastN (Altschul et al. 1997) tool in the Blast v.2.4.0+ package (http://blast.ncbi.nlm.nih.gov) with a matched standard “e-value = 1e-10 and matching ratio = 100%”. The program PANTHER v.10 (Mi et al. 2016) was used to perform the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, and the threshold was set to a P-value < 0.05 after the Bonferroni correction. The bioinformatics analysis workflow is summarized in Fig. S2.
mRNA differential expression and functional analysis
After the mapping, the programs Cufflinks, Cuffmerge, and Cuffdiff in the Cufflinks software package v.2.1.1 (Trapnell et al. 2012) were used to assemble the mRNA clean reads and calculate the gene expression levels based on the fragments per kilobase of exon per million fragments mapped (FPKM) values. First, we performed the mRNA transcript assembly and created a “merged transcripts.gtf” for all ovarian or endometrial samples from one species using the programs Cufflinks and Cuffmerge. Then, the program Cuffdiff was used to calculate the level of gene expression and perform differential expression analyses between the two species (i.e., ovary/endometrium: European mouflon vs. Finnsheep) or two types of tissues (i.e., European mouflon: ovary vs. endometrium). The differentially expressed genes (DEGs) were defined as genes that showed |log2(fold change)| ≥ 2 and Padj (i.e. adjusted P-value) ≤ 0.01 in the comparisons. Furthermore, we performed GO and KEGG pathway enrichment analyses of functional genes associated with the differentially expressed mRNAs using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tools v.6.8 (Huang et al. 2009). The enriched GO terms were considered significant at an adjusted P-value < 0.05. A word cloud was created for the GO terms and pathways using an online Wordcloud generator (https://www.jasondavies.com/wordcloud/).
miRNA quantification and differential expression analysis
The miRDeep2 v.0.0.7 software package (Friedländer et al. 2012) was used to predict the putative precursor miRNA sequences, and precursor miRNAs with 10 or fewer reads and miRDeep2 scores < 5 were removed. Then, the filtered miRNAs were aligned to the miRNA database miRBase v.21 (http://www.mirbase.org/) to identify mature miRNAs that contain sheep miRNAs (i.e., miRNAs deposited in the miRBase of O. aries), conserved miRNAs (i.e., miRNAs matching those in other mammals in miRBase with fewer than four mismatches), and novel miRNAs (remaining miRNAs).
The differentially expressed ovarian and endometrial miRNAs (including the sheep and conserved miRNAs) between European mouflon and Finnsheep and between the two types of tissues (ovary vs. endometrium) in European mouflon were identified using the DESeq v.1.22.0 program (Anders and Huber 2010). By applying the threshold values of Padj (i.e., adjusted P-value) ≤ 0.05 and |log2(fold change)| ≥ 1, the remaining miRNAs considered to be upregulated or downregulated in one of the two species or types of tissues were used in the following mRNA–miRNA integrated analyses.
miRNA–mRNA regulatory network analysis
The program TargetScan v.7.0 (Agarwal et al. 2015) was used to predict the binding of the differentially expressed miRNAs to their putative targets, i.e., the 5′ seed regions (2–8 nt) of the miRNAs complementary to the conserved 8- and 7-mer sites in the 3′-untranslated region of the mRNA (Lewis et al. 2005). miRNAs typically function in post-transcriptional regulation by suppressing or silencing specific target genes. Thus, we searched for inverse correlations between the expression levels of the miRNAs and their predicted target genes, i.e., between the upregulated miRNAs and downregulated mRNAs, and vice versa. To narrow the list of potential miRNA–target interactions, the putative target genes were overlapped with the DEGs in the mRNA profiling data. Then, target genes with a context++ score percentile lower than 50 were removed. Furthermore, the upregulated and downregulated miRNAs and their target genes were included in the network visualization analysis using the program Cytoscape v.3.4.0 (Shannon et al. 2003). Only the top 10 downregulated miRNAs were considered core miRNAs and used to build the main regulatory network. Furthermore, GO and pathway enrichment analyses of the miRNA target genes were conducted using DAVID tools v.6.8 (Huang et al. 2009). GO terms and pathways with an adjusted P-value < 0.05 were considered significantly enriched. A flowchart of the mRNA–miRNA integrated analysis is provided in Fig. S1.
To confirm the credibility of the inferred miRNA and target gene pairs, we searched the miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/; Chou et al. 2016) database and previous studies for differentially expressed miRNAs, predicted target genes, and miRNA–gene pairs consistent with those identified in this study.
Results and Discussion
mRNA and miRNA profiling
In total, we generated approximately 127.1 Gb mRNA and 21.3 Gb miRNA raw data. After filtering, we obtained a large number of high-quality reads: 1083.6 million mRNA and 56.8 million miRNA reads in European mouflon, and 934.6 million mRNA and 127.8 million miRNA reads in Finnsheep. Most high-quality mRNA and miRNAs have been mapped to the O. aries reference genome Oar_v.4.0 (Table S2). Regarding the mRNA expression, more than 89% of the genes were expressed at <50 FPKM and only <1.1% of the genes were expressed at >500 FPKM in all four tissue-species combinations (Table S3). Altogether, 23 557 common genes were expressed in the ovaries and 19 208 common genes were expressed in the endometria (Table S3).
In the de novo assembly of the European mouflon transcriptome, 1083.6 million clean mRNA reads were assembled into 844 129 unigenes, with an N50 length of 700 bp and an average length of 562 bp (Table 1). The observed length of N50 is comparable to that reported in domestic sheep (e.g., 508–1482 bp) (Yue et al. 2015; Zhang et al. 2015), thus highlighting the high quality of the assembled European mouflon transcriptome. Based on the transcriptome, we identified 5739, 5831, 5897, and 5981 annotated DEGs in the four comparisons between the ovary and endometrium tissues (M2-OA vs. M2-EA, M2-OA vs. M2-EB, M2-OB vs. M2-EA, and M2-OB vs. M2-EB) in European mouflon, respectively. However, when the analyses were performed using the sheep reference genome (Oar v.4.0), we found far fewer DEGs (e.g., 1925 DEGs for M2-O-B vs. M2-E-B). Despite the possible implication of using the O. aries genome as a reference, the observation suggests that the de novo approach is accurate and that the quality of the de novo assembly is good.
We identified 373 mature miRNAs in the ovaries and 192 mature miRNAs in the endometria, including 89 and 59 sheep miRNAs, 174 and 104 conserved miRNAs, and 110 and 29 novel miRNAs in the two tissues, respectively (Table S4). High levels of expression were observed in the sheep miRNAs, followed by conserved miRNAs and novel miRNAs (Table S4). Of the 122 novel miRNAs (Table S5), 20 miRNAs were also identified as novel miRNAs and validated in our previous study (Hu et al. 2016). The chromosomal distribution of the sheep and conserved miRNAs indicated the presence of large numbers of miRNAs on chromosomes 18 and X, whereas none of the miRNAs were expressed on chromosome 8 based on the ovarian data and chromosomes 8, 17, and 25 based on the endometrium data (Table S6).
mRNAs and miRNAs showing the greatest expression
Of the genes exhibiting the greatest expression (FPKM > 3000) in ovary, 7 genes in the Finnsheep and 15 genes in the European mouflon encode ribosomal protein (Fig. 1a and Table S7). In the endometrium, only 1 top gene identified in the Finnsheep and 22 top genes identified in the European mouflon encode ribosomal protein involved in the ribosome pathway (Fig. 1b and Table S7). The ribosome pathway has been reported to be among the most significant pathways involved in early embryogenesis (Berres et al. 2017). The observed large number of ribosome family genes in both tissues of European mouflon could be one explanation for the low reproductive success of the wild sheep (i.e., seasonal estrus, 1.5 years of age to sexual maturation and one lamb per litter; Cugnasse et al. 1985; Garel et al. 2005) compared to the prolificacy of domestic sheep, including Finnsheep (i.e., out-of-season breeding, 6 months of age to sexual maturation and three lambs per litter on average; Maijala 1996; Li et al. 2011; Mullen and Hanrahan 2014). Moreover, the observed smaller number of ribosome family genes expressed at a higher level in Finnsheep than in European mouflon is likely due to the evolution of reproductive traits between the two species during the domestication and long-term artificial selection processes. Additionally, this observation could be related to the environment of the mouflon being harsher than the domestic environment of the Finnsheep; in such a harsh environment, it may be beneficial to have fewer offspring.

Number of genes exhibiting the greatest expression (FPKM > 3000) in the Finnsheep and European mouflon ovaries (a) and endometria (b), and their overlap with genes in the ribosome pathway
Of the 10 most highly expressed ovarian miRNAs, 7 (miR-10b, miR-143, miR-26a, miR-27b, let-7f, miR-191, and miR-22-3p) miRNAs are common between European mouflon and Finnsheep (Table S8). Of the endometrial miRNAs, three (miR-10b, miR-100, and miR-34c-5p) miRNAs are highly expressed in both species (Table S8). We also observed tissue- and species-specific miRNAs (Table S9), which could possibly be associated with particular functions. For instance, miR-181a, which is specifically and abundantly expressed in the Finnsheep ovary, was demonstrated to be associated with pregnancy complications and was more highly expressed in normal term than in pre-term delivery (Mayor-Lynn et al. 2011). Additionally, miR-186, which is specifically and abundantly expressed in the European mouflon endometrium, was observed to be more highly expressed in endometrial cancer (Zhou et al. 2008).
Differentially expressed mRNAs and miRNAs
In total, 192 and 1065 DEGs responsible for ovarian and endometrial mRNAs were identified between the two species, respectively. Of these DEGs, 149 ovarian and 492 endometrial DEGs were upregulated in European mouflon, while 43 ovarian and 573 endometrial DEGs were upregulated in Finnsheep (Tables S10 and S11). Additionally, we identified 53 and 5 differentially expressed ovarian and endometrial miRNAs between the two species, respectively (Table 2). Based on the differentially expressed mRNAs and miRNAs in the tissues, heat maps were constructed (Fig. 2) and showed distinct expression patterns between European mouflon and Finnsheep, indicating that the evolution of the gene- and post-transcriptional regulation could have contributed to the improved reproductive performance during the domestication process.

Heat maps of differentially expressed mRNAs and miRNAs between Finnsheep and European mouflon. a Heat map showing the expression intensity of 1065 differentially expressed genes (DEGs) based on a comparison of endometria from the two species. b Heat map showing the expression intensity of 192 DEGs based on a comparison of ovaries from the two species. c Heat map showing the expression intensity of 5 differentially expressed miRNAs based on a comparison of endometria from the two species. d Heat map showing the expression intensity of 53 differentially expressed miRNAs based on a comparison of ovaries from the two species. Heat maps were generated using the R heat map package v.1.0.8 (http://cran.r-project.org/web/packages/pheatmap/), which is based on the k-means clustering algorithm and the Boolean values to determine if the rows (i.e., mRNAs or miRNAs) should be clustered. The important genes associated with the reproductive traits such as early mature, fecundity, non-seasonal breeding, and total lambs born are labeled in the figure
Of the upregulated genes in the ovary and endometrium tissues from the Finnsheep, 4 genes (CYP19, PTGS2, FST, and PAPSS2) are associated with early maturity, 14 genes (EREG, INHBA, SERPINA5, SPP1, LIPG, PTGS2, AREG, TDGF1, FST, C4BPA, SERPINE2, TRPV3, LDLR, and TINAGL1) are associated with fecundity, and two genes (CYP19 and SERPINE2) are associated with non-seasonal breeding. In addition, we compared the 192 ovarian DEGs with the QTLs for the reproductive traits in the Sheep QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/OA/), and found 8 genes (NME9, LRRIQ4, SPATC1L, PRDM15, PEX5L, COL6A6, RARRES1, and SPOCK1; Table S12) overlapped with previously identified QTLs associated with traits such as reproductive seasonality and total lambs born. Similarly, we found 73 endometrial DEGs that overlapped with the reported QTLs for the reproductive seasonality, and 1 gene, i.e., RASL11B, was overlapped with the QTL chromosome 6 as follows: 68295117–68445447 for the total lambs born trait (Table S13).
GO and KEGG enrichments of differentially expressed mRNAs
In the functional enrichment analysis of the 192 ovarian DEGs between European mouflon and Finnsheep, the top enriched GO categories were mainly relevant to reproduction, including fertilization, reproductive developmental process, reproductive process, oogenesis, and ovarian follicle development (Fig. S3a and Table S14). Fertilization refers to the fusion of gametes that initiates the development of a new individual organism (Merriam-Webster 2017). During the process, the ZP proteins (ZP1 to ZP4) in the zona pellucida function to surround the egg, bind the sperm and mediate species-selective sperm–oocyte interactions (Suzuki et al. 2015; Avella et al. 2016). ZP2 peptide beads have been shown to markedly inhibit the fertilization of ovulated eggs, prevent sperm binding in mice and humans (Gupta et al. 2012; Avella et al. 2016) and induce infertility in female mice (Avella et al. 2016). ASTL, which is involved in the negative regulation of fertilization by the ZP2 and ZP4 genes, plays a definitive role in ensuring monospermic fertilization by encoding ovastacin, which is a cortical granule protease (Burkart et al. 2012). Thus, the significantly upregulated expression of the ASTL, ZP2, ZP3, and ZP4 genes in the European mouflon ovary (Table S10) may indicate that these genes play a role in the low fertility of wild sheep compared to the high prolificacy of domestic sheep (Fig. 3).

Functional roles of the main mRNAs and miRNAs in the endometrium and ovary of Ovis species. Ovarian and endometrial network of differentially expressed mRNAs and top 10 Finnsheep downregulated (i.e., European mouflon upregulated) miRNAs and target genes are shown, along with their functional roles associated with reproduction (bold type). Regulation and alteration of these functional roles are responsible for the evolution of high prolificacy traits. Red and blue arrows indicate downregulation and upregulation of the reproductive organs, respectively
The GO terms of the reproductive development process and ovarian follicle development included the genes EREG, INHBA, SERPINA5, and SPP1, which are significantly upregulated in Finnsheep (Tables S10 and S14). EREG is a member of the epidermal growth factor family, which plays an important role in mediating the luteinizing hormone-induced ovulation process in preovulatory follicles (Amsterdam 2010; Kim et al. 2011). INHBA plays an essential role in early oocyte and follicular development (Thomas et al. 2003) and has a significant effect on the litter size in sheep (Leyhe et al. 1994) and goat (Zi et al. 2012). SERPINA5, which encodes a serine protease inhibitor, may be involved in follicular growth and atresia in the bovine ovarian follicle (Hayashi et al. 2011). SPP1 is located on sheep chromosome 6 close to the high prolificacy gene FecB in sheep (Montgomery et al. 1993; Montgomery et al. 1994). Additionally, the genes AMH, TDRD5, SOHLH1, and NOBOX are upregulated in European mouflon (Tables S10 and S14). AMH is a central influencing factor in gonad development, and could inhibit germ cell proliferation and differentiation in both sexes (Durlinger et al. 2002). Recent clinical data have indicated that serum AMH level is a useful indicator of ovarian aging and premature ovarian failure in women (de Vet et al. 2002; Alipour et al. 2015). The TDRD5 protein is an evolutionarily conserved protein involved in germ cell development. Injection of TDRD5-deficient round spermatids into oocytes result in fertile offspring (Yabuta et al. 2011). SOHLH1 is a critical regulator of oogenesis and can downregulate NOBOX, which functions to disrupt folliculogenesis (Pangas et al. 2006). Therefore, our results suggest that the upregulated expression of these genes (i.e., EREG, INHBA, SERPINA5, and SPP1 in Finnsheep, and AMH, SOHLH1, TDRD5, and NOBOX in European mouflon) is most likely associated with the hyper-prolificacy of domestic sheep (e.g., Finnsheep) and the single lambing of wild sheep (e.g., European mouflon; Fig. 3). We also observed other top GO categories related to immune processes such as the humoral immune response and humoral immune response mediated by circulating immunoglobulin (Table S14). The KEGG analysis of the 192 ovarian DEGs identified three significant (adjusted P-value < 0.05) pathways, i.e., ovarian steroidogenesis, complement and coagulation cascades, and MAPK signaling pathways (Table S14). The MAPK (i.e., p38αMAPK) pathway has been shown to play an essential role in female reproduction, particularly in follicular development (Hu et al. 2017) (Fig. 3).
In the functional enrichment of the endometrial DEGs between the wild and domestic sheep, the GO terms of the 492 upregulated genes (RARRES2, LTBP1, and TGFB3) in European mouflon were most significantly enriched in the developmental process, such as anatomical structure development (Fig. S3b and Table 3), whereas the top enriched GO terms of the 573 upregulated genes (PTGS2, LDLR, and SREBF1) in Finnsheep included biosynthetic and metabolic processes, such as lipid metabolic process (Fig. S3b and Table 3). An early study investigating the bovine endometrial transcriptome indicated an earlier shift from proliferation to metabolism at early diestrus in the cow (Mesquita et al. 2015). Cell proliferation was enriched in the small follicle endometrium, whereas biosynthetic and metabolic processes were enriched in the large follicle endometrium, which presented higher proliferative activity in the luminal epithelium, glandular epithelium, and stroma than in the small follicle endometrium (Mesquita et al. 2015). Additionally, the large follicle endometrium is an active endometrial phenotype that is associated with the optimal uterine environment (Miller and Moore 1976; Mesquita et al. 2015). Thus, Finnsheep could have a larger follicle endometrium and better receptivity of the uterus than European mouflon (Fig. 3).
In the KEGG analysis of the 1065 endometrial DEGs, the top enriched pathways were enriched in the extracellular matrix (ECM)–receptor interaction, steroid biosynthesis, and biosynthetic and metabolic processes (Table S15). The ECM, which is a complex matrix of biological macromolecules, can transfer signals to cells via surface receptors during cell adhesion and other various signaling transduction pathways. Additionally, the ECM can send signals to the cytoplasm and nucleus to influence gene expression or cellular activities (Alldinger et al. 2006). Thus, adhesion and connection mechanisms could play important roles in the endometrium at the molecular level, and different cell adhesion may occur between wild and domestic sheep. In addition, the differentially expressed mRNAs between the ovarian and endometrial tissues from the European mouflon and their functional analysis based on the reference genome and de novo approaches are presented in the Supplementary Results and Tables S16–S18.
Post-transcriptional regulatory network of miRNAs and target genes
We obtained 100, 64, 17, and 51 target genes for the up- and downregulated miRNAs in particular tissues (Table 4). In the main downregulated network of the Finnsheep ovary, which comprised 10 downregulated miRNAs and 57 upregulated target genes (Fig. 4a), several important miRNAs and functional genes were involved in the reproductive process. For instance, miR-136, which exhibited the largest fold change (Table 2), has been shown to be downregulated in term labor women compared with that in pre-term labor women by real-time quantitative reverse transcription-PCR (qRT-PCR) (Montenegro et al. 2009). The genes EREG and INHBA, which are co-regulated by three miRNAs (miR-136, miR-374a, and miR-9-5p), were identified as important genes responsible for the hyper-prolificacy of Finnsheep in the above GO enrichment analysis of ovarian mRNAs (Table 2). Additionally, four miRNAs (miR-101, miR-3959-5p, miR-411a-5p, and miR-9-5p) targeted PIAS1 and five miRNAs (miR-708-3p, miR-101, miR-374a, miR-3959-5p, and miR-9-5p) targeted UBE3A. Both genes modulate androgen receptor (AR)-dependent transactivation (Nishida and Yasuda 2002; Ramamoorthy and Nawaz 2008). AR is well-known for its roles in male reproduction and has been recently revealed to be essential for normal female fertility, including follicle health, development, and ovulation (Walters et al. 2010). Moreover, four miRNAs (miR-136, miR-101, miR-148a, and miR-9-5p) co-regulated KITLG, which is a strong candidate gene related to litter size in goats (An et al. 2015).

Main miRNA–gene regulatory networks. a Ovarian network of top 10 Finnsheep downregulated (i.e., European mouflon upregulated) miRNAs and 57 target genes. b Ovarian network of top 10 European mouflon downregulated (i.e., Finnsheep upregulated) miRNAs and 64 target genes. c Endometrial network of 1 Finnsheep upregulated (i.e., European mouflon downregulated) miRNAs and 17 target genes. d Endometrial network of 4 Finnsheep downregulated (i.e., European mouflon upregulated) miRNAs and 51 target genes. Large and small ellipses represent miRNAs and their target genes, respectively. The important miRNAs, genes, and miRNA–gene pairs discussed in the text are marked with red color
The main downregulated network of the European mouflon ovary comprises 10 downregulated miRNAs that co-regulate 64 upregulated target genes (Fig. 4b). In the network, miR-370-5p, which exhibited the second largest fold change (Table 2), was reported to be differentially expressed between the ovaries of sheep producing single lamb and two lambs, and plays a vital role in sheep follicular development and litter size regulation (Qi et al. 2015). Additionally, miR-1306 exhibits a lower expression level in the ovaries of monotocous goats than that in ovaries of polytocous goats (An et al. 2016).
Regarding the endometrial networks, Finnsheep had 1 upregulated miRNA with 17 downregulated target genes and 4 downregulated miRNAs with 51 upregulated target genes, while European mouflon had inverse regulation patterns (Fig. 4c, d). In the upregulated network of European mouflon (Fig. 4d), miR-92a targeted the gene TNFAIP6, which is a key catalyst in the formation of the cumulus extracellular matrix and is indispensable for female fertility (Fülöp et al. 2003). Additionally, miR-100 was observed to be highly expressed in monotocous goats (An et al. 2016). In addition, other identified miRNAs and target genes (Figs. S4 and S5) may also influence the reproductive traits of European mouflon and Finnsheep (see Supplementary Results).
In the GO enrichment analysis of the genes that were targeted by the differentially expressed ovarian and endometrial miRNAs between Finnsheep and European mouflon (Supplementary Results and Tables S19–S22), the 64 upregulated genes targeted by 23 downregulated miRNAs in the ovary of Finnsheep were significantly enriched in the GO categories of the reproductive process and regulation of developmental process (Table S19). Altogether, our integrated network analysis suggested that the post-transcriptional downregulation of miRNAs and the corresponding upregulated expression of the target genes in the ovary of Finnsheep most likely account for the evolution of reproduction traits from the monotocous wild European mouflon to the highly prolific domestic Finnsheep (Fig. 3).
Regarding the 58 differentially expressed miRNAs, 210 predicted target genes and 1025 miRNA–gene pairs between Finnsheep and European mouflon, 27 miRNAs, 52 genes, and 67 miRNA–gene pairs have been reported in previous studies (Table S23), thus supporting our findings. Further studies involving a larger panel of species/breeds and sample sizes using qRT-PCR could help validate the relevance of the miRNAs, their target genes, and the miRNA–gene pairs revealed in this study.
Conclusion
To our knowledge, this study provides the first mRNA–miRNA integrated profiling analysis of ovarian and endometrial tissues in wild and domestic sheep. By comparing the RNA-Seq data of the ovaries and endometria between European mouflon and Finnsheep, we identified differentially expressed mRNAs and miRNAs. The functional enrichment and miRNA–mRNA regulatory network analyses revealed novel genes and miRNAs associated with reproductive traits in wild and domestic sheep. Our results provide new insight into the molecular mechanisms and evolution of the high prolificacy traits in the Ovis species (Fig. 3). Additionally, the large amount of mRNA and miRNA data and the de novo transcriptome of European mouflon generated here provide valuable resources for studies investigating transcript functions associated with ovine reproduction traits under domestication and selection.
Data archiving
The RNA-sequencing data reported in this article have been deposited in the Nucleotide and Sequence Read Archive databases (https://www.ncbi.nlm.nih.gov). The Illumina raw sequence reads appear in SRA under accession SRP142554 and under BioProject PRJNA451237.
References
Abdoli R, Zamani P, Deljou A, Rezvan H (2013) Association of BMPR-1B and GDF9 genes polymorphisms and secondary protein structure changes with reproduction traits in Mehraban ewes. Gene 524:296–303
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4:e05005
Alipour F, Rasekhjahromi A, Maalhagh M, Sobhanian S, Hosseinpoor M (2015) Comparison of specificity and sensitivity of AMH and FSH in diagnosis of premature ovarian failure. Dis Markers 2015:585604
Alldinger S,Gröters S,Miao Q,Fonfara S,Kremmer E,Baumgärtner W,(2006) Roles of an extracellular matrix (ECM) receptor and ECM processing enzymes in demyelinating canine distemper encephalitis Dtsch Tierarztl Wochenschr 113:151–152, 154–156
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488
Amsterdam A (2010) Novel role of growth factors in ovary function. Harefuah 149:789–793
An X, Song Y, Hou J, Li G, Zhao H, Wang J et al. (2016) Identification and profiling of microRNAs in the ovaries of polytocous and monotocous goats during estrus. Theriogenology 85:769–780
An XP, Hou JX, Gao TY, Lei YN, Song YX, Wang JG et al. (2015) Association analysis between variants in KITLG gene and litter size in goats. Gene 558:126–130
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:1
Avella MA, Baibakov BA, Jimenez-Movilla M, Sadusky AB, Dean J (2016) ZP2 peptide beads select human sperm in vitro, decoy mouse sperm in vivo, and provide reversible contraception. Sci Transl Med 8:336ra60
Barrett EL, Moore AJ, Moore PJ (2009) Diet and social conditions during sexual maturation have unpredictable influences on female life history trade-offs. J Evol Biol 22:571–581
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Berezikov E (2011) Evolution of microRNA diversity and regulation in animals. Nat Rev Genet 12:846–860
Berres ME, Garic A, Flentke GR, Smith SM (2017) Transcriptome profiling identifies ribosome biogenesis as a target of alcohol teratogenicity and vulnerability during early embryogenesis. PLoS ONE 12:e0169351
Bodin L, Di Pasquale E, Fabre S, Bontoux M, Monget P, Persani L et al. (2007) A novel mutation in the bone morphogenetic protein 15 gene causing defective protein secretion is associated with both increased ovulation rate and sterility in Lacaune sheep. Endocrinology 148:393–400
Bronson FH (2009) Climate change and seasonal reproduction in mammals. Philos Trans R Soc Lond B Biol Sci 364:3331–3340
Burkart AD, Xiong B, Baibakov B, Jiménez-Movilla M, Dean J (2012) Ovastacin, a cortical granule protease, cleaves ZP2 in the zona pellucida to prevent polyspermy. J Cell Biol 197:37–44
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH et al. (2016) miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44:D239–D247
Clark EL, Bush SJ, McCulloch MEB, Farguhar IL, Young R, Lefevre L et al. (2017) A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet 13:e1006997
Cugnasse J-M, Garcia M, Veyrac T (1985) Contribution à l’étude de la biologie de la reproduction du mouflon (Ovis ammon musimon), par examen post-mortem, dans le massif du Caroux-Espinouse. Bull Mens Natl Chass 89:33–35
de Vet A, Laven JS, de Jong FH, Themmen AP, Fauser BC (2002) Antimüllerian hormone serum levels: a putative marker for ovarian aging. Fertil Steril 77:357–362
Di R, He J, Song S, Tian D, Liu Q, Liang X et al. (2014) Characterization and comparative profiling of ovarian microRNAs during ovine anestrus and the breeding season. BMC Genomics 15:899
Durlinger AL, Visser JA, Themmen AP (2002) Regulation of ovarian function: the role of anti-Müllerian hormone. Reproduction 124:601–609
Fazleabas AT, Strakova Z (2002) Endometrial function: cell specific changes in the uterine environment. Mol Cell Endocrinol 186:143–147
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52
Fülöp C, Szántó S, Mukhopadhyay D, Bárdos T, Kamath RV, Rugg MS et al. (2003) Impaired cumulus mucification and female sterility in tumor necrosis factor-induced protein-6 deficient mice. Development 130:2253–2261
Garel M, Cugnasse J-M, Gaillard J-M, Loison A, Gibert P, Douvre P et al. (2005) Reproductive output of female mouflon (Ovis gmelini musimon × Ovis sp.): a comparative analysis. J Zool 266:65–71
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Gupta SK, Bhandari B, Shrestha A, Biswal BK, Palaniappan C, Malhotra SS et al. (2012) Mammalian zona pellucida glycoproteins: structure and function during fertilization. Cell Tissue Res 349:665–678
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Hanrahan JP, Gregan SM, Mulsant P, Mullen M, Davis GH, Powell R et al. (2004) Mutations in the genes for oocyte derived growth factors GDF9 and BMP15 are associated with both increased ovulation rate and sterility in Cambridge and Belclare sheep (Ovis aries). Biol Reprod 70:900–909
Hayashi KG, Ushizawa K, Hosoe M, Takahashi T (2011) Differential gene expression of serine protease inhibitors in bovine ovarian follicle: possible involvement in follicular growth and atresia. Reprod Biol Endocrinol 9:72
Hiendleder S, Kaupe B, Wassmuth R, Janke A (2002) Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies. Proc Biol Sci 269:893–904
Hu XJ, Pokharel K, Peippo J, Ghanem N, Zhaboyev I, Kantanen J et al. (2016) Identification and characterization of miRNAs in the ovaries of a highly prolific sheep breed. Anim Genet 47:234–239
Hu S, Rao M, Lei H, Wu Y, Wang Y, Ke D et al. (2017) Expression patterns of p38αMAPK during follicular development in the ovaries of neonatal rats. Acta Histochem 119:538–542
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T et al. (2014) The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344:1168–1173
Kim K, Lee H, Threadgill DW, Lee D (2011) Epiregulin-dependent amphiregulin expression and ERBB2 signaling are involved in luteinizing hormone-induced paracrine signaling pathways in mouse ovary. Biochem Biophys Res Commun 405:319–324
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:1
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20
Leyhe B, Hiendleder S, Jaeger C, Wassmuth R (1994) Pronounced differences in the frequency of TaqI beta A-inhibin alleles between sheep breeds with different reproductive performance. Anim Genet 25:41–43
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li MH, Strandén I, Tiirikka T, Sevón-Aimonen ML, Kantanen JA (2011) Comparison of approaches to estimate the inbreeding coefficient and pairwise relatedness using genomic and pedigree data in a sheep population. PLoS ONE 6:e26256
Lincoln GA (1989) Seasonal cycles in testicular activity in Mouflon, Soay sheep and domesticated breeds of sheep: breeding seasons modified by domestication. Zool J Linn Soc 95:137–147
Liu B, Li J, Cairns MJ (2014) Identifying miRNAs, targets and functions. Brief Bioinform 15:1–19
Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC (2008) The evolution and functional diversification of animal microRNA genes. Cell Res 18:985–996
Lv FH, Peng WF, Yang J, Zhao YX, Li WR, Liu MJ et al. (2015) Mitogenomic meta-analysis identifies two phases of migration in the history of eastern Eurasian sheep. Mol Biol Evol 32:2515–2533
Maijala K (1996) The Finnsheep. In: Fahmy MH (ed) Prolific Sheep. CAB International, Wallingford, Oxon, UK, p 10–46
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Mayor-Lynn K, Toloubeydokhti T, Cruz AC, Chegini N (2011) Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor. Reprod Sci 18:46–56
McDaneld TG (2009) MicroRNA: mechanism of gene regulation and application to livestock. J Anim Sci 87:21–28
Mesquita FS, Ramos RS, Pugliesi G, Andrade SCS, Hoeck VVan, Langbeen A et al. (2015) The receptive endometrial transcriptomic signature indicates an earlier shift from proliferation to metabolism at early diestrus in the cow. Biol Reprod 93:52
Merriam-Webster Online (2017) Fertilization. Available at www.merriam-webster.com/dictionary/fertilization, last accessed August 3, 2017
Mi H, Poudel S, Muruganuian A, Casagrande JT, Thomas PD (2016) PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44:D336–D342
Miao X, Qin QL (2015) Genome-wide transcriptome analysis of mRNAs and microRNAs in Dorset and Small Tail Han sheep to explore the regulation of fecundity. Mol Cell Endocrinol 402:32–42
Miller BG, Moore NW (1976) Effects of progesterone and oestradiol on RNA and protein metabolism in the genital tract and on survival of embryos in the ovariectomized ewe. Aust J Biol Sci 29:565–573
Montenegro D, Romero R, Kim SS, Tarca AL, Draghici S, Kusanovic JP et al. (2009) Expression patterns of microRNAs in the chorioamniotic membranes: a role for microRNAs in human pregnancy and parturition. J Pathol 217:113–121
Montgomery GW, Crawford AM, Penty JM, Dodds KG, Ede AJ, Henry HM et al. (1993) The ovine Booroola fecundity gene (FecB) is linked to markers from a region of human chromosome 4q. Nat Genet 4:410–414
Montgomery GW, Lord EA, Penty JM, Dodds KG, Broad TE, Cambridge L et al. (1994) The Booroola fecundity (FecB) gene maps to sheep chromosome 6. Genomics 22:148–153
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Mullen MP, Hanrahan JP (2014) Direct evidence on the contribution of a missense mutation in GDF9 to variation in ovulation rate of Finnsheep. PLoS ONE 9:e95251
Nishida T, Yasuda H (2002) PIAS1 and PIASxα function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J Biol Chem 277:41311–41317
Pangas SA, Choi Y, Ballow DJ, Zhao Y, Westphal H, Matzuk MM et al. (2006) Oogenesis requires germ cell-specific transcriptional regulators Sohlh1 and Lhx8. Proc Natl Acad Sci USA 103:8090–8095
Peters H, McNatty KP (1980) The Ovary: A Correlation of Structure and Function in Mammals. University of California Press, Berkeley and Los Angeles, California, p 201
Pokharel K, Peippo J, Andersson G, Li MH, Kantanen J (2015) Transcriptome profiling of Finnsheep ovaries during out-of-season breeding period. Agric Food Sci 24:1–9
Pokharel K, Peippo J, Honkatukia M, Seppälä A, Rautiainen J, Ghanem N et al. (2018) Integrated ovarian mRNA and miRNA transcriptome profiling characterizes the genetic basis of prolificacy traits in sheep (Ovis aries). BMC Genomics 19:104
Qi YX, Liu XF, Zhang P, He XL, Xing YM, Dalai et al. (2015) Differentially expressed microRNAs screening between ovaries of sheep producing single lamb and twins. Sci Agric Sin 48:2039–2048.
Ramamoorthy S, Nawaz Z (2008) E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal 6:e006
Rasali DP, Shrestha JNB, Crow GH (2006) Development of composite sheep breeds in the world: a review. J Anim Sci 86:1–24
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Santiago-Morenoa J, López-Sebastiána A, González-Bulnesa A, Gómez-Bruneta A, Chemineaub P (2000) Seasonal changes in ovulatory activity, plasma prolactin, and melatonin concentrations, in Mouflon (Ovis gmelini musimon) and Manchega (Ovis aries) ewes. Reprod Nutr Dev 40:421–430
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Suravajhala P, Kogelman LJA, Kandarmideen HN (2016) Multi-omic data integration and analysis using systems genomics approaches: methods and applications in animal production, health and welfare. Genet Sel Evol 48:38
Suzuki K, Tatebe N, Kojima S, Hamano A, Orita M, Yonezawa N (2015) The hinge region of bovine zona pellucida glycoprotein ZP3 is involved in the formation of the sperm-binding active ZP3/ZP4 complex. Biomolecules 5:3339–3353
Thomas FH, Walters KA, Telfer EE (2003) How to make a good oocyte: an update on in-vitro models to study follicle regulation. Hum Reprod Update 9:541–555
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Vinet A, Drouilhet L, Bodin L, Mulsant P, Fabre S, Phocas F (2012) Genetic control of multiple births in low ovulating mammalian species. Mamm Genome 23:727–740
Walker RS, Gurven M, Burger O, Hamil MJ (2008) The trade-off between number and size of offspring in humans and other primates. Proc Biol Sci 275:827–833
Walters KA, Simanainen U, Handelsman DJ (2010) Molecular insights into androgen actions in male and female reproductive function from androgen receptor knockout models. Hum Reprod Update 16:543–558
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Warriach HM, McGill DM, Bush RD, Wynn PC, Chohan KR (2015) A review of recent developments in buffalo reproduction—a review. Asian-Australas J Anim Sci 28:451–455
Wira CR, Rodriguez-Garcia M, Patel MV (2015) The role of sex hormones in immune protection of the female reproductive tract. Nat Rev Immunol 15:217–230
Yabuta Y, Ohta H, Abe T, Kurimoto K, Chuma S, Saitou M (2011) TDRD5 is required for retrotransposon silencing, chromatoid body assembly, and spermiogenesis in mice. J Cell Biol 192:781–795
Yue YJ, Liu JB, Yang M, Han JL, Guo TT, Guo J et al. (2015) De novo assembly and characterization of skin transcriptome using RNAseq in sheep (Ovis aries). Genet Mol Res 14:1371–1384
Zhang C, Wang G, Hou L, Ji Z, Wang J (2015) De novo assembly and characterization of the skeletal muscle transcriptome of sheep using Illumina paired-end sequencing. Biotechnol Lett 37:1747–1756
Zhou B, Yang L, Sun Q, Cong R, Gu H, Tang N et al. (2008) Cigarette smoking and the risk of endometrial cancer: a meta-analysis. Am J Med 121:501–508
Zi XD, Xu HW, Wang Y (2012) Variation in sequences and mRNA expression levels of inhibin subunits α (INHA) and βA (INHBA) genes between prolific and nonprolific goat breeds. Mol Reprod Dev 79:238
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (grants Nos. 31661143014 and 31272413), the Climate Genomics for Farm Animal Adaptation (ClimGen) project, the Academy of Finland (grants Nos. 250633 and 256077), and the National Transgenic Breeding Project of China (2014ZX0800952B), the External Cooperation Program of Chinese Academy of Sciences (152111KYSB20150010), and the Taishan Scholars Program of Shandong Province (No. ts201511085). The authors thank CSC (www.csc.fi) for the computing infrastructure and Finnish Functional Genomics Centre supported by University of Turku, Åbo Akademi University and Biocenter Finland.
Author contributions
M.-H.L. and J.K. conceived and designed the project; J.K., M.H., and J.P. collected the samples; K.P. implemented the de novo transcriptome assembly; X.L., Y.-H.C., X.-J.H., and Z.-H.C. analyzed the data; M.-H.L., X.L., Y.-H.C., and J.Y. wrote the paper with contribution from S.-S.X. All authors reviewed and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Li, X., Cao, YH. et al. Comparative mRNA and miRNA expression in European mouflon (Ovis musimon) and sheep (Ovis aries) provides novel insights into the genetic mechanisms for female reproductive success. Heredity 122, 172–186 (2019). https://doi.org/10.1038/s41437-018-0090-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0090-1
- Springer Nature Switzerland AG
This article is cited by
-
Structural variant landscapes reveal convergent signatures of evolution in sheep and goats
Genome Biology (2024)
-
Variations of follicular fluid extracellular vesicles miRNAs content in relation to development stage and season in buffalo
Scientific Reports (2022)
-
Genome-wide identification and comparison of differentially expressed profiles of miRNAs and lncRNAs with associated ceRNA networks in the gonads of Chinese soft-shelled turtle, Pelodiscus sinensis
BMC Genomics (2020)
-
Seasonal effects on miRNA and transcriptomic profile of oocytes and follicular cells in buffalo (Bubalus bubalis)
Scientific Reports (2020)