
Abstract
Yes-associated protein (YAP) is one of major key effectors of the Hippo pathway and the mechanism supporting abnormal YAP expression in Anaplastic thyroid carcinoma (ATC) remains to be characterized. Here, we identified ubiquitin carboxyl terminal hydrolase L3 (UCHL3) as a bona fide deubiquitylase of YAP in ATC. UCHL3 stabilized YAP in a deubiquitylation activity-dependent manner. UCHL3 depletion significantly decreased ATC progression, stem-like and metastasis, and increased cell sensitivity to chemotherapy. Depletion of UCHL3 decreased the YAP protein level and the expression of YAP/TEAD target genes in ATC. UCHL3 promoter analysis revealed that TEAD4, through which YAP bind to DNA, activated UCHL3 transcription by binding to the promoter of UCHL3. In general, our results demonstrated that UCHL3 plays a pivotal role in stabilizing YAP, which in turn facilitates tumorigenesis in ATC, suggesting that UCHL3 may prove to be a potential target for the treatment of ATC.
Similar content being viewed by others


Introduction
Anaplastic thyroid carcinoma, also known as undifferentiated carcinoma, continues to rank as one of the deadliest diseases worldwide and carries a very poor prognosis [1,2,3]. ATC is a rare but extremely aggressive type of thyroid cancer derived from follicular cells of the thyroid gland [4, 5]. Although ATC accounts for only 2–5% of all thyroid tumors, it is responsible for 33–50% of all thyroid cancer-related deaths [6, 7]. Currently, there exists no effective or standard therapy to cure or prolong the survival of ATC patients due to its inherent resistance to both radioactive iodine and conventional chemotherapy [5, 8]. Thus, it is important to understand the underlying mechanisms during the initiation and progression of ATC.
The Hippo tumor suppressor pathway is highly conserved in mammals which was originally known to restrict organ size [9]. Accumulating studies have also demonstrated that the Hippo pathway plays a prominent role in regulating tumorigenesis [10]. Yes-associated protein (YAP) is one of major key effectors of the Hippo pathway. As transcriptional co-activators, YAP mediates the biological functions of the Hippo pathway by regulating gene transcription [11]. Dysregulation of YAP has been observed in many types of cancers, including breast, colon, liver, lung, ovary, and others [12, 13]. High YAP activity promotes cell proliferation, survival, migration, and invasion. Its activation is important for cell to escape contact inhibition and anoikis and to support anchorage-independent growth [14]. In addition, the activity of YAP is associated with chemoresistance of cancer. Breast cancer cells with high YAP activity demonstrated resistance to drugs such as doxorubicin, 5-fluorouracil, and taxol [15].
The activity of YAP is mainly regulated by the MST1/2-Lats1/2 kinase cascade by directly phosphorylating YAP on multiple sites, resulting in interaction with 14-3-3 protein and cytoplasmic retention [16, 17]. Besides the mechanisms regulating its phosphorylation and localization, YAP can be controlled by other post-translational modifications such as ubiquitination. For instance, Fbxw7 and PARK2 regulates YAP protein stability by targeting YAP for ubiquitination and proteasomal degradation [18, 19]; SHARPIN and RNF187 promotes YAP degradation via inducing YAP K48-dependent polyubiquitination [20, 21]. Deubiquitinating enzymes (DUB) are also involved in regulation of YAP in human cancers. USP9X, USP47 and USP10 were reported as potent YAP/TAZ-activating DUBs, which directly interact with and stabilizes YAP/TAZ by reverting their proteolytic ubiquitination [22,23,24]. However, the DUB responsible for YAP deubiquitination and stabilization in ATC is largely unknown. DUBs modulate protein fate in a specific or selective manner. Targeting DUBs can modulate the signaling status of the cell by modifying specific “Key Aspects” of the pathological pathways. DUBs that target oncogenic proteins may be targeted by compounds to inhibit their activity. Development and design of an enzyme selective/specific inhibitor is easier than designing and developing an enzyme activator (due to competitive inhibition and modeling of substrates) [25]. Development of DUB selective/specific inhibitors is emerging as attractive targets for cancer treatment. Therefore, we concentrated our focus on DUBs involved in YAP activation.
In the present study, we first screened a DUB siRNA library and identified UCHL3 as a potent deubiquitinase responsible for YAP deubiquitination and stabilization in ATC. We found that UCHL3 promoted cancer progression, stem-like and metastasis in response to chemotherapy drugs treatment through YAP. Overall, our study demonstrated that UCHL3 was a novel deubiquitinating enzyme of YAP, suggesting that it may prove to be a potential target for the treatment of ATC.
Results
UCHL3 stabilizes YAP through the deubiquitylation activity
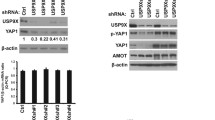
We previously reported that YAP promotes the proliferation of ATC cells [26]. To further dissection of the YAP pathway in ATC, we performed RNA sequencing to further approach the function of YAP in ATC (Fig. S1A). Gene ontology analysis indicated that down-regulated genes induced by YAP knockdown were significantly enriched in cell adhesion and mitosis, while the upregulated genes were enriched in trans−synaptic signaling and type I interferon signaling (Fig. S1B, C). GSVA analysis revealed that YAP is associated with thyroid cancer, cell cycle and metabolism of tumor (Fig. S1D). We then utilized a siRNA screen library to identify the deubiquitinating enzymes responsible for YAP deubiquitylation and stabilization in ATC. Four non-overlapping siRNA mixtures specific for each of the DUBs were transfected into CAL-62 cells. It was found that silencing UCHL3 markedly decreased YAP (Fig. 1A). We tested the expression of UCHL3 in 10 thyroid cancer cell lines (TPC-1, B-cpap, IHH4, FTC133, TT, CAL-62, 8505c, KHM-5M, 8305c, BHT-101) and immortalized normal thyroid epithelial cell line (Nthy-ori3-1), we observed an upregulation tendency of UCHL3 in ATC cell lines, especially in CAL-62 and KHM-5M cells (Fig. S2A). Then we depleted UCHL3 with two non-overlapping siRNAs separately in CAL-62 and KHM-5M cells to further validate the function of UCHL3 in regulating YAP protein level (Fig. 1B). Consistently, ectopic expression of UCHL3 profoundly upregulated YAP in a dose-dependent manner. While the catalytically inactive mutant C95A (UCHL3C95A) lost its ability to upregulate YAP, suggesting that UCHL3 regulated YAP in a DUB activity-dependent manner (Fig. 1C). RT-PCR analysis indicated that UCHL3 did not change the mRNA abundance of YAP (Fig. 1D). We examined the expression of YAP target genes (CTGF, CYR61 and ANKRD1) and found that depletion of UCHL3 decreased the transcripts of CTGF, CYR61 and ANKRD1 (Fig. S2B, C). In addition, we measured YAP/TEAD-luciferase reporter gene activity by UCHL3 depletion to determine whether UCHL3 depletion affected the YAP transcriptional activity. Our results showed that UCHL3 depletion decreased the YAP/TEAD-luciferase reporter gene activity in CAL-62 cells (Fig. 1E). TAZ is a YAP homology with overlapping function, we then examined whether UCHL3 could affect TAZ expression and ubiquitination. It is found that the expression and ubiquitination of TAZ were not affected by UCHL3 (Fig. S3A). We detected the possibility that UCHL3 in deubiquitylating YAP, and found that UCHL3 deletion decreased the YAP protein level, and this effect could be reversed by addition of the proteasome inhibitor MG132 or overexpression of UCHL3-WT, but not its catalytically inactive mutant UCHL3C95A (Fig. 1F, G). We then treated cells with the protein synthesis inhibitor cycloheximide to determine whether UCHL3 could affect YAP stability. It was found that the stability of YAP was decreased in cells depleted of UCHL3 (Fig. 1H). In cells overexpressing UCHL3-WT, but not UCHL3C95A, the half-life of YAP was prolonged (Fig. 1I). IHC analysis of two tissue microarrays (TMA) indicated that UCHL3 and YAP were both upregulated in ATC samples (Fig. S4A, B). Besides, we observed a positive correlation between UCHL3 and YAP protein levels in human thyroid cancer samples (Fig. 1J). Taken together, these results indicated that UCHL3 was a regulator of Hippo signaling pathway by stabilizing YAP.

A The siRNAs specific to each deubiquitinating enzyme were transfected into CAL-62 cells. After 48 h, cells were lysed and the YAP protein level was analyzed by Western blot. B UCHL3 depletion decreased YAP protein level without affecting mRNA expression of YAP. C Increasing amounts of UCHL3 WT or C95A were transfected into CAL-62 cells and YAP expression was detected. D UCHL3 did not change the mRNA abundance of YAP. E UCHL3 depletion decreased YAP-luciferase activity. CAL-62 cells were transfected with SiUCHL3 or SiControl together with YAP-luciferase reporter plasmid. Luciferase activity was measured 48 h after transfection. F CAL-62 cells transfected with the indicated siRNA were treated with or without the proteasome inhibitor MG132 (10 µM, 6 h), and then proteins were analyzed. G UCHL3 WT or C95A was introduced into CAL-62 cells together with UCHL3 siRNA. YAP levels were measured. H CAL-62 cells transfected with UCHL3 siRNA were treated with cycloheximide (10 µg ml−1), and collected at the indicated times for western blot. I CAL-62 cells were transfected with UCHL3 plasmid or siRNA, and YAP mRNA levels were measured. J YAP correlated with UCHL3 in thyroid cancer samples. Commercially available tissue microarray slides (Alenabio, China) were purchased for IHC analysis. K An immunofluorescence assay demonstrated that UCHL3 and YAP at least partially colocalized in CAL-62 and KHM-5M cells. L Co-IP assay revealed an association between endogenous UCHL3 and YAP in CAL-62 cells. CAL-62 cells were harvested with RIPA lysis buffer. Co-IP was performed using antibody as indicated. M CAL-62 cells transfected with Myc-UCHL3 were lysed and the lysates were incubated with GST-YAP or GST protein. The interacted UCHL3 was detected by western blot. N YAP and UCHL3 domain structure and deletion mutants used in the study. O UCHL3 interacted with the WW domain of YAP. P C terminal of UCHL3 physically interacted with YAP. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by unpaired, two-tailed Student’s t tests.
UCHL3 interacts with YAP
We then performed immunofluorescence assay assess cellular localization of UCHL3 and YAP. The results of immunostaining demonstrated that YAP and UCHL3 colocalized both in the nuclear and cytosol of ATC cells (Fig. 1K). Our co-immunoprecipitation (Co-IP) experiment demonstrated that endogenous UCHL3 could coimmunoprecipitate with endogenous YAP (Fig. 1L). GST-pull-down assay showed that UCHL3 interacted with YAP in vitro in a manner that is independent of its DUB activity (Fig. 1M). Additionally, deletion analysis demonstrated that the C terminal of UCHL3 physically interacted with the WW domain of YAP (Fig. 1N–P). We further deleted the WW domain of YAP (YAP-ΔWW) and detected its stability. It is found that mutation in the WW domain in YAP de-stabilized the protein and UCHL3 could not prolong the half-life of YAP-ΔWW (Fig. S3B). These findings indicated that UCHL3 and YAP form an intact complex.
UCHL3 deubiquitylates YAP
As UCHL3 is a deubiquitylase, we went on to examine the possibility that UCHL3 deubiquitylates YAP. Depletion of UCHL3 significantly increased the level of ubiquitinated-YAP (Fig. 2A). We then performed endogenous ubiquitination assay and detected the endogenous ubiquitin on YAP. As shown in Fig. 2B, overexpression of UCHL3 decreased the endogenous ubiquitin on YAP. Consistently, ectopic expression of UCHL3-WT, but not UCHL3 C95A, markedly decreased YAP ubiquitylation in cells both in vivo and in vitro (Fig. 2C, D). We then treated CAL-62 cells with TCID, a small molecule inhibitor for UCHL3, finding that similar to UCHL3 siRNA, inhibition of UCHL3 by TCID enhanced YAP ubiquitination in a dose-dependent manner (Fig. 2E). Furthermore, UCHL3 decreased the YAP ubiquitylation induced by the E3 ligase SHARPIN (Fig. 2F). In vivo deubiquitylation assays also showed that UCHL3 removed the ubiquitin chain of YAP in a time- and dose-dependent manner (Fig. 2G). We further performed ubiquitination assay with a series of ubiquitin mutants to investigate which type of ubiquitin chain of YAP was deubiquitylated by UCHL3. It was found that UCHL3 efficiently removed the K11 and K48-linked ubiquitin chain on YAP (Fig. 2H). Taken together, UCHL3 proved to be a specific DUB, which de-polyubiquitylated and stabilized YAP.

A CAL-62 cells transfected with the indicated siRNA were treated with MG132 for 6 h before collection. YAP was immunoprecipitated with anti-YAP and immunoblotted with anti-HA. B Immunoblotting was used to detect the endogenous ubiquitination of YAP in CAL-62 cells transfected with Myc-UCHL3. C Immunoblotting was used to detect the ubiquitination of YAP in 293T cells co-transfected with Flag-YAP, HA-Ubiquitin and Myc-UCHL3 (wild-type or C95A). D Wild-type UCHL3, but not UCHL3 C94A possesses DUB activity toward polyubiquitinated YAP in vitro. Ubiquitinated YAP was purified from HEK293T cells transfected with HA-Ub, and Flag-YAP plasmids using anti-Flag affinity purification method. The ubiquitinated YAP was incubated with GST-tagged wild-type UCHL3, and UCHL3 C95A proteins in 20 µl reaction buffer containing at 37 °C for 2 h. The reactions were terminated by boiling in 1X SDS sample buffer and analyzed using IB with anti-HA antibodies. E Protein lysates were collected from CAL-62 cells treated with increasing amounts of TCID for 48 h. YAP was immunoprecipitated with anti-YAP and immunoblotted with anti-HA. Cells were exposed to MG132 (10 μM) for 6 h before harvest. F YAP ubiquitylation was analyzed in cells transfected with E3 SHARPIN together with UCHL3 or not. G UCHL3 removed the ubiquitin chain of YAP in a time- and dose-dependent manner. H HA-WT, K6, K11, K27, K29, K33, K48 or K63 Ub were co-transfected with Flag-YAP and Myc-UCHL3 into HEK293T cells. After treatment with 10 μM MG132 for 6 h, cell lysates were subjected to ubiquitination assay and the ubiquitination level of YAP detected by HA antibody. I CAL-62 cells transfected with empty vector or UCHL3 were treated with MG132 for 6 h before collection. YAP was immunoprecipitated with anti-YAP and immunoblotted with anti-K48.
UCHL3 is a direct transcriptional target of YAP/TEAD
Regulation of UCHL3 transcription is not yet well understood. It was found in our study that YAP depletion in ATC cells significantly decreased the UCHL3 mRNA level (Fig. S5A), suggesting that UCHL3 may be a transcriptional target of YAP/TEAD. We then used PROMO database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) and JASPAR database (http://jaspar.genereg.net/) to analyze the human UCHL3-promoter nucleotide sequence, and found two putative TEAD4 binding motifs (Fig. S5B, C). We then constructed and transfected a series of luciferase reporter plasmids flanked with truncated or mutated UCHL3 promoter sequences into CAL-62 cells for a luciferase reporter assay. The result of serial deletion suggested that the binding site R2 (−409 to −398 bp) was essential to TEAD4-induced expression of luciferase reporter. In addition, site-directed mutation of UCHL3 also indicated that the binding site R2 was indispensable for TEAD4 binding and the transcription of luciferase reporter (Fig. S5D, E). Furthermore, the chromatin immunoprecipitation (ChIP) assay demonstrated that TEAD4 directly bound to the putative sites of UCHL3 in ATC cell lines (Fig. S5F). These findings demonstrated that UCHL3 was a direct transcriptional target of YAP/TEAD.
UCHL3 promotes ATC progression, metastasis and stem-like properties
We next examined the role of UCHL3 in regulating ATC progression. Our results demonstrated that depletion of UCHL3 significantly decreased cell proliferation and migration. Depletion of UCHL3 decreased cell proliferation and increased the population in G1 phases, indicating that UCHL3 may regulate G1 to S transition in ATC cells (Fig. 3A, B). The results of clone formation assay revealed that UCHL3 depletion significantly decreased the clone formation capability in CAL-62 and KHM-5M cells (Fig. 3C). Consistently, EdU incorporation assay indicated that DNA synthesis was inhibited in CAL-62 and KHM-5M cells treated with UCHL3 siRNAs (Fig. 3D, E). Furthermore, depletion of UCHL3 significantly decreased the cell migration capacity as revealed by wound healing and transwell assays (Fig. 3F–H).

A UCHL3 depletion inhibited the cell proliferation in ATC cells. B UCHL3 depletion induced G1 cell cycle arrest in ATC cells. C UCHL3 depletion decreased the clone formation capability of anaplastic thyroid cancer cells. D, E Representative images of EdU assay of ATC cells. F, G Wound-healing assay of anaplastic thyroid cancer cells. H Transwell migration assay of ATC cells. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by unpaired, two-tailed Student’s t tests.
As ATC is an undifferentiated carcinoma, we then examined the role of UCHL3 in anaplastic thyroid cancer stemness characteristics. It was found that UCHL3 depletion significantly reduced the oncosphere formation of CAL-62 and KHM-5M cells (Fig. 4A, B). Notably, UCHL3 depletion significantly reduced the expression of pluripotent transcription factors Sox2, Nanog, KLF4 and Oct4 compared with control cells (Fig. 4C) and decreased ALDH activity of ATC cells (Fig. 4D). Moreover, CAL-62 cells expressing shUCHL3 displayed an attenuated tumor-initiation capacity in NOD-SCID mice compared with control cells (Fig. 4E). Overall, UCHL3 silencing abrogates the tumorigenic capacity of anaplastic thyroid cancer stem cells.

A, B Sphere formation assay of UCHL3-knockdown cells and control cells. C Depletion of UCHL3 decreased the expression of pluripotent factors in ATC cells as assessed by quantitative real-time PCR. D Depletion of UCHL3 decreased the ALDH activity of ATC cells. E Tumorigenic cell frequency in UCHL3-knockdown cells and control cells was determined with limiting dilution assays. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by unpaired, two-tailed Student’s t tests.
To determine the mechanism of UCHL3 in regulating ATC cell proliferation and migration by stabilizing YAP, we performed rescue experiments by ectopic expressing YAP or UCHL3C95A in UCHL3 knockdown CAL-62 cells. CCK8 assay indicated that overexpression of YAP largely recovered the proliferation rate of CAL-62 cells (Fig. 5A). Increased YAP expression reversed the clone formation ability of CAL-62 cells (Fig. 5B). Consistently, YAP overexpression also facilitated the DNA synthesis in CAL-62 cells depleted with UCHL3 (Fig. 5C). Wound healing and transwell assays showed that the suppressive function induced by UCHL3 depletion was largely reversed by YAP overexpression (Fig. 5D, E). But the UCHL3C95A lost the ability of promoting proliferation and migration. Knockdown of UCHL3 significantly inhibited tumor growth in vivo, while the restoration of YAP expression abolished the inhibition induced by UCHL3 depletion (Fig. 5F–H). We further used a tail vein injection mouse model for in vivo metastasis evaluation. Depletion of UCHL3, observably inhibited lung metastasis in mice, which effect could be rescued by YAP overexpression (Fig. 5H, I). Consistently, UCHL3 overexpression promote ATC tumor growth and metastasis in vivo, and this effect could be abolished by knockdown of YAP (Fig. S6A). Taken together, these results indicate that UCHL3 promotes ATC progression through YAP.

A Cell proliferation assay of CAL-62. B Clone formation assay of CAL-62. C Representative images of EdU assay of CAL-62. D Wound-healing assay of CAL-62. E Transwell migration assay of CAL-62. F Overexpression of YAP in UCHL3-knockdown cells partly recovered tumor growth in vivo. 1 × 106 CAL-62 cells transfected with indicated plasmids were injected to the right dorsal flank of each mouse (n = 6). Tumor sizes were measured every 3 days until the end of the experiment. The data are presented as the mean ± SEM. G Western blot images of UCHL3 and YAP. H Representative images of immunohistochemical staining for Ki67, UCHL3 and YAP. I, J UCHL3 depletion suppressed the lung metastasis of ATC in mice. 0.5 × 106 ATC cells were intravenously injected into each mouse through the tail vein (n = 6). The lungs were harvested 4 weeks after injection. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by unpaired, two-tailed Student’s t tests.
TCID inhibits the growth and metastasis of ATC cells
We also tested the effect of pharmacological UCHL3 inhibitor TCID (0, 5, 10 μM) on the proliferation and migration of anaplastic thyroid cancer cells. As shown in Fig. 6, inhibition of UCHL3 by TCID caused a dose-dependent suppression on the growth and metastasis of ATC cells (Fig. 6A–E). Moreover, we examined whether TCID affected the ATC growth and metastasis in vivo and found that treatment of TCID resulted in an obvious decrease in tumor growth and metastasis in nude mice (Fig. 6F–H). To further determine whether the anti-tumor and anti-metastatic effects of the TCID are mediated by UCHL3 and YAP, we constructed UCHL3 and YAP knockdown cells. It is found that TCID imposed minimal effect on the growth and metastasis of UCHL3 and YAP knockdown cells (Fig. S6B, C). Consistent with our previous experiments, inhibition of UCHL3 by TCID significantly decreased YAP protein level in ATC cells (Fig. 6I). Co-treatment of cycloheximide and TCID decreased the level of endogenous YAP more rapidly (Fig. 6J). The effect of TCID on YAP was mainly dependent on UCHL3, as TCID had no effect on the YAP protein level in UCHL3-knockdown cells, and depletion of UCHL1 did not affect YAP protein levels (Fig. 6K).

A TCID inhibited cell proliferation in ATC cells. B TCID decreased clone formation capability of ATC cells. C Representative images of EdU assay of ATC cells. D Wound-healing assay of ATC cells. E Transwell migration assay of ATC cells. F Nude mice with tumors formed by CAL-62 cells were treated with vehicle control or TCID intraperitoneally (10 mg/kg, 3 times each week). G Representative images of immunohistochemical staining for Ki67 and YAP. H TCID suppressed the lung metastasis of ATC in mice. 0.5 × 106 ATC cells were intravenously injected into each mouse through the tail vein (n = 6). Nude mice were treated with vehicle control or TCID intraperitoneally (10 mg/kg, 3 times each week). The lungs were harvested 4 weeks after injection. I Protein lysates were collected from the indicated ATC cells treated with the indicated concentrations of TCID for 48 h. Western blot assay was used to detect the expression of UCHL3 and YAP proteins. J CAL-62 cells treated with TCID or DMSO were treated with cycloheximide for various lengths of time. Western blot assay was used to detect YAP and UCHL3 protein levels. K TCID increased K48-linked polyubiquitination of YAP protein. L TCID decreased YAP protein levels via UCHL3. CAL-62 cells transfected with the indicated siRNA were treated with TCID, and then proteins were analyzed. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by unpaired, two-tailed Student’s t tests.
UCHL3 decreases the response of ATC cells to chemotherapy through YAP
Doxorubicin is the most commonly used chemotherapy drug for ATC. YAP activation has been shown to play a role in chemoresistance in various types of malignancies including ovarian cancer, breast cancer, and hepatocellular carcinoma [27,28,29,30,31,32,33]. Knowing that UCHL3 could stabilize YAP, we examined whether inhibition of UCHL3 affected cell response to doxorubicin. As shown in Fig. 7, inhibition of UCHL3 by siRNA or TCID sensitized CAL- 62 cells to chemotherapy drugs treatment (Fig. 7A, B), and the effect induced by UCHL3 depletion could be abolished by YAP reconstitution (Fig. 7A). Consistently, CAL- 62 cells with overexpression of WT UCHL3 developed resistance against doxorubicin treatment, while cells overexpressing catalytically inactive mutant UCHL3 showed no such effect (Fig. 7C). The same results were observed in KHM-5M cells (Fig. 7G, F). To confirm the anti-chemotherapy function of UCHL3 in vivo, CAL-62 cells overexpressing an empty vector, UCHL3, YAP or shRNA targeting YAP were xenografted into nude mice. It was found that tumor growth of the control CAL-62 cells was significantly reduced by doxorubicin treatment, while tumor cells overexpressing UCHL3 or YAP grew more rapidly than the control cells on doxorubicin treatment, and depletion of YAP abolished the chemoresistance effect induced by UCHL3 overexpression (Fig. 7G). Furthermore, administration of TCID and doxorubicin in combination significantly inhibited tumor growth in vivo than administration of either TCID or doxorubicin alone (Fig. 7H).

A CAL-62 cells expressing indicated constructs were treated with doxorubicin, and cell survival was determined. B CAL-62 cells were co-treated with TCID and doxorubicin, and cell survival was determined. C CAL-62 cells expressing indicated constructs were treated with doxorubicin, and cell survival was determined. D KHM-5M cells expressing indicated constructs were treated with doxorubicin, and cell survival was determined. E KHM-5M cells were co-treated with TCID and doxorubicin, and cell survival was determined. F KHM-5M cells expressing indicated constructs were treated with doxorubicin, and cell survival was determined. G In vivo xenografts generated from CAL-62 cells expressing an empty vector, UCHL3, YAP or YAP-targeting shRNA and treated with doxorubicin. 1 × 106 CAL-62 cells were injected to the right dorsal flank of each mouse (n = 6). After the tumors reached ~50 mm3, the mice were treated with doxorubicin (3 mg/kg/day, diluted into injectable NaCL with a final concentration of 50 µl) delivered intratumorly. H CAL-62 xenografts treated with doxorubicin alone or in combination with TCID. 1 × 106 CAL-62 cells were injected to the right dorsal flank of each mouse (n = 6). After the tumors reached ~50 mm3, mice were treated with doxorubicin (3 mg/kg/day, diluted into injectable NaCl with a final concentration of 50 µl) delivered intratumorly. The animals were then treated with the vehicle or TCID intraperitoneally (10 mg/kg/day, 3 times each week). Tumor sizes were measured every 3 days until the end of the experiment. The data are presented as the mean ± SEM. Results shown are representative of three independent experiments. Data are represented as mean ± SD of biological triplicates. *p value < 0.05; **p value < 0.01; ***p value < 0.001 by two-way ANOVA tests.
Discussion
ATC is a rare, highly aggressive malignant tumor. There exists no effective or standard therapy for the treatment of anaplastic thyroid cancer. Therefore, it is an urgent issue to explore the underlying molecular mechanisms involved in the initiation and progression of anaplastic thyroid cancer. More novel candidate targets are needed to improve the treatment decisions. Our findings demonstrate that UCHL3 is a YAP DUB that promotes ATC progression through stabilizing YAP, which in turn activates UCHL3 transcription (Fig. S7). Targeting UCHL3/YAP may enable promising therapeutic approaches for the treatment of ATC patients.
In our previous study, we observed that YAP is essential for the progression of ATC tumors [26]. YAP is a major factor of the Hippo tumor suppressor pathway. The expression of YAP is associated with cell proliferation, cancer metastasis, poor prognosis, resistance to chemotherapy, and greater possibility of relapse [34, 35], making it attractive therapeutic targets in cancer. However, the mechanisms underlying YAP overexpression or activation in malignant cells have not been well defined. The Hippo pathway, and specifically the YAP/TEAD transcriptional complex, has been shown to be a promising target for the treatment for various cancers: verteporfin was identified from a small library of FDA-approved compounds, which could block the interaction between YAP and TEADs to suppress tumor growth in mice [36]. While verteporfin has also been reported to have proteotoxic effects [37], suggesting that this compound may not translate well to the clinic. The compounds targeting YAP/TEADs activity only blocks the biological functions mediated by YAP/TEADs transcriptional complex, they are unable to block the biological effects of YAP independent of TEADs. To date, no small molecule compounds targeting Hippo pathway have progressed to the clinic [38]. To systematically evaluate the impact of the individual members of the DUB family in YAP deubiquitination and promoting tumor progression, we have screened a DUB siRNA library and conducted unbiased siRNA screening by monitoring the levels of YAP and identified several candidate DUBs. In addition to UCHL3, we also noticed a few other DUBs (USP10 [24], USP47 [23] and EIF3H [39]) with potential effect on the YAP/TAZ signaling pathway, which were recently found to revert the proteolytic ubiquitination of YAP. However, siOTUB1 [40], siOTUB2 [41] or siMINDY1 [42] imposed minimal effect on YAP in our screening systems. The seemingly contradicting results may arise from the variation of cellular context in different cell lines.
UCHL3 is characterized by dual hydrolase specificity for DUB and deneddylating activities, and belongs to the UCH family [43]. It is involved in multiple biological processes, including insulin signaling, preimplantation embryo development, fertilization, DNA repair, osteoblast differentiation and tumor progression [44,45,46,47,48,49]. UCHL3 has emerged as a tumor suppressor in prostate cancer. Its overexpression suppressed epithelial-to-mesenchymal transition (EMT), and inhibited cancer cell invasion and metastasis [50]. The oncogenic role of UCHL3 has also been reported in recent studies: UCHL3 activates NF-κB signaling pathway by deubiquitinating and stabilizing TRAF2, thus inducing overactive inflammatory response of ovarian cancer [51]; UCHL3 promotes pancreatic cancer progression in a FOXM1-dependent manner [52]; UCHL3 functions to promote tumor growth and cancer stemness via enhancing AhR protein stabilization. The UCHL3 inhibitor TCID or depletion of UCHL3 by its specific shRNAs induced AhR degradation in cells and inhibited tumor cell proliferation [53].
In the present study, we identified UCHL3 as a potent DUB responsible for YAP deubiquitination and stabilization in ATC. First, UCHL3 and YAP interacted with each other directly. Co-IP analysis identified the association between YAP and UCHL3. Binding assays demonstrated that UCHL3 interacted with the WW domain of YAP in a manner independent of its DUB activity. Second, UCHL3 decreased YAP polyubiquitination and promotes YAP protein stabilization in a DUB activity-dependent manner. UCHL3 deletion markedly decreased YAP levels, and this effect could be reversed by addition of the proteasome inhibitor MG132 or overexpression of UCHL3-WT, but not its catalytically inactive mutant UCHL3C95A. Upon inhibition of protein synthesis by cycloheximide, UCHL3 depletion significantly decreased the half-life time of YAP protein. Ectopic expression of UCHL3-WT, but not UCHL3C95A, markedly decreased YAP ubiquitylation both in vivo and in vitro. In vivo deubiquitylation assays showed that UCHL3 directly removed the ubiquitin chain of YAP in a time- and dose-dependent manner. To further analyze the underlying mechanisms, a series of mutant ubiquitin were used to identify the linkage of ubiquitin chain. We observed that UCHL3 significantly decreased K6- and K48-linked polyubiquitination from YAP. As polyubiquitination through K48 of Ub generally results in proteasomal degradation [54, 55], UCHL3 may maintain the stability of YAP by removing the K48-linked ubiquitin chain from YAP protein. Previous studies indicated that SHARPIN could promote YAP degradation via inducing YAP K48-dependent polyubiquitination. It was found in our study that UCHL3 decreased the YAP ubiquitylation induced by the E3 ligase SHARPIN. Finally, UCHL3 could promote cancer progression, metastasis, stem-like properties and chemoresistance of ATC through YAP. Depletion of UCHL3 significantly decreased cancer progression and increased cell sensitivity to chemotherapy. In addition, the restoration of YAP expression abolished the effects induced by UCHL3 depletion. We further examined TCID, a small molecular inhibitor of UCHL3, to verify our findings. As expected, TCID treatment caused a dose-dependent suppression on the proliferation and migration of ATC cells and elevated cell sensitivity to chemotherapy. Correspondingly, TCID treatment reduced the YAP protein level and inhibited the deubiquitinase activity of UCHL3 against YAP. Cotreatment with TCID and CHX significantly shortened the half-life of YAP, suggesting that TCID, like UCHL3 knockdown, promotes YAP ubiquitination and degradation.
In conclusion, we demonstrated that UCHL3 is a YAP DUB that promotes tumor growth, metastasis and tumor stem-like properties through stabilizing YAP though its deubiquitylation activity. Our findings provide new insights into the roles of UCHL3 in Hippo signaling pathway, and suggest that UCHL3 may prove to be a potential target for the treatment of ATC.
Materials and methods
Cell culture
The human ATC cell lines CAL-62, KHM-5M and human embryonic kidney HEK293T cells were obtained from the Chinese Academy of Sciences (Shanghai, China). All cell lines were authenticated by the cell banks with short tandem repeat analysis. CAL-62 and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, 41965, Life Technologies), and KHM-5M cells were maintained in RPMI 1640 (42401, Life Technologies). All media were supplemented with 10% fetal bovine serum (FBS, Gibco, Life Technologies, 10270). All cells were cultured at 37 °C in an atmosphere of 5% CO2.
Plasmids and RNA inference
The YAP, wild-type (WT) UCHL3 and its inactive mutant plasmids were obtained from Hanbio Biotechnology Co., Ltd. (Shanghai, China). The HA-K6, -K11, -K27, -K29, -K33, -K48, -K63, and -Ub plasmids were acquired from Addgene. Small interfering RNAs (siRNAs) targeting UCHL3 (siRNA-1: 5′-CAGCAUAGCUUGUCAAUAA-3′; 5′-GCAAUUCGUUGAUGUAUAU-3′) were synthesized by Genepharma (Shanghai, China).
Establishment of stable expression cell lines
To generate lentivirus, lentivirus vectors (pCDH-UCHL3-Puro, pCDH-YAP-Puro, PLVX-shUCHL3-Puro, PLVX-shYAP-Puro, 10 μg) and lentiviral components (10 μg psPAX2 and 5 μg pMD2.G) were co-transfected into 2 × 106 293T cells in a 10 cm2 dish using Lipofectamine 3000. Lentivirus-containing supernatants from 48 and 72 h post-transfection were collected to infect ATC cells. The stable cell lines were selected using 2 μg/ml puromycin for 3 days for two passages. Puromycin (1 μg/ml) was used to maintain the cells.
RNA extraction and qRT-PCR analysis
The total RNA was extracted from the cancer cells using the RNeasy plus mini kits (Qiagen, Germany). Reverse transcription was performed using the PrimeScript RT Master Mix (Takara, Japan). qRT-PCR was performed using the SYBR green mix (Toyobo, Japan) with the CFX96TM Real-time PCR Detection System (Bio-Rad, USA). The 2−ΔΔCt method was used to calculate the relative expression. GAPDH was used for internal control. All assays were performed in triplicates. Primers were listed as follows: GAPDH (forward: 5′-ACGGGAAGCTTGTCATCAAT-3′, reverse: 5′-TGGACTCCACGACGTACTCA-3′); YAP (forward: 5′-TAGCCCTGCGTAGCCAGTTA-3′; reverse: 5′-TCATGCTTAGTCCACTGTCTGT-3′); ANKRD1 (forward: 5′-AGAACTGTGCTGGGAAGACG-3′; reverse: 5′-GCCATGCCTTCAAAATGCCA-3′); CTGF (forward: 5′-CTCGCGGCTTACCGACTG-3′; reverse: 5′-GGCTCTGCTTCTCTAGCCTG-3′); CYR61 (forward: 5′-AGCAGCCTGAAAAAGGGCAA-3′; reverse: 5′-AGCCTGTAGAAGGGAAACGC-3′).
Luciferase assay
The YAP/TEAD-luciferase reporter plasmid, Renilla plasmid and UCHL3 siRNA were transfected together into CAL-62 cells. Luciferase activity was detected using the Dual-Luciferase Reporter kit (Promega, Germany).
Co-immunoprecipitation assay
Cells were lysed with NP-40 lysis buffer containing a cocktail of protease inhibitors. The total cell lysis was precleared with rabbit IgG for 2 h and subsequently immunoprecipitated with indicated antibody at 4 °C overnight. Protein A/G PLUS-Agarose beads (Santa Cruz) were then added to the lysates and incubated at 4 °C for 2 h. The immunocomplexes were washed with lysis buffer three times and separated by SDS-PAGE. Immunoblotting was performed following standard procedures.
GST pulldown assays
GST fusion proteins bound to glutathione–Sepharose 4B beads (Samgon Biotech, China) were incubated with cell lysates at 4 °C for 2 h. Then the beads were washed with GST binding buffer. The bound proteins were separated by SDS-PAGE, followed by Western blot with indicated antibodies.
Chromatin immunoprecipitation (ChIP) assay
Magna ChIP-seq ™ Chromatin Immunoprecipitation Kit (Millipore, Billerica, USA) was used for detecting protein-chromatin interactions. Briefly, the cells were fixed with formaldehyde, sonicated and incubated with the target protein. The cross-linked DNA fragments were then released from the co-precipitated complexes, purified, and amplified by PCR. Sequences of primers amplifying the UCHL3 promoter regions were as follows: Region 1 (R1), sense: 5′-CTGAGACATGCTGCCCTCAA-3′, antisense: 5′-TCCACCGTCTGGTTTATATGGT-3′; Region 2 (R2), sense: 5′-GGTTCAGTCCAGGAATCCACTT′-3′; antisense: 5′-GGAATTACGGAGGAAGGGGG-3′; Distant Region, sense: 5′-TGCCAAAAGGCCACGTTTAG-3; antisense: 5′-CTTCCTGCTGCATGGGTTTG-3′.
Protein stability assay
To measure the half-life of YAP, cells were treated with 100 µM protein synthesis inhibitor cycloheximide (Sigma-Aldrich) for indicated times. Western blot was performed to measure protein levels.
In vitro deubiquitination assay
HEK293T cells were transfected with empty vector or Flag-YAP and HA-Ub plasmids. Forty-eight hours after transfection, ubiquitinated YAP proteins were immunoprecipitated with anti-FLAG M2 affinity gels (Sigma-Aldrich). The ubiquitinated YAP proteins were incubated with bacterially purified GST or GST-UCHL3 (wild-type or C95A) proteins in deubiquitination buffer (50 mM NaCl, 50 mM Tris-HCl, 10 mM dithiothreitol, 1 mM EDTA, 5% glycerol, pH 8.0) at 37 °C for 2 h. After the reaction, reaction mix was supplemented with SDS to a final concentration of 2%, boiled at 95 °C to dissociate all protein-protein interactions, diluted 10 to 20 times via the deubiquitination buffer, and then immunoprecipitated by Flag beads before the beads are boiled again and assessed for the HA-ubiquitin level by WB.
In vivo deubiquitination assay
In vivo deubiquitination assay was performed in HEK293T and ATC cells. HEK293T cells were transfected with HA-Ub, Flag-YAP, Myc-UCHL3 or Myc-UCHL3 C95A plasmid as indicated for 48 h. After treatment with 10 μM MG132 for 6 h, cells were lysed directly in 10 mM Tris-HCl buffer containing 2% SDS and boiled. Flag-YAP was immunoprecipitated with anti-Flag and immunoblotted with anti-HA. In ATC cells, HA-Ub plasmid were co-transfected with UCHL3 siRNAs into CAL-62 cells. And the experimental procedures were performed as described above.
Western blot analysis
Cells were lysed with RIPA extraction reagent (Beyotime, China) supplemented with protease inhibitors (Sigma-Aldrich, USA). Total protein was separated using 10–12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.45 μm PVDF membrane (Millipore, USA). Primary antibodies were YAP (Proteintech, 13584-1-AP), UCHL3 (Proteintech, 12384-1-AP), HA (Proteintech, 51064-2-AP), Myc (Proteintech, 60003-2-Ig), GAPDH (Proteintech, 60004-1-Ig) antibodies. Bands were visualized using an enhanced chemiluminescence (ECL) kit (Boster, China) and detected by ChemiDoc XRS + Imaging System (Bio-Rad).
Cell proliferation and cell viability analysis
The cell proliferation rate was detected using Cell Counting Kit-8 (CCK8) assay at indicated time points according to the manufacturer’s instructions. Briefly, cultured CAL-62 and KHM-5M cells were digested and 2 × 103 cells were seeded in 96-well culture plates. CCK8 solution reagent was added to each well and incubated for 1.5 h at 37 °C. The absorbance of the colored solution was measured at a wavelength of 450 nm with a spectrophotometer. For clone formation assay, 1000 cells were seeded into 6-well plates. After 14-day incubation, cells were fixed with 4% paraformaldehyde and visualized by 0.5% crystal violet staining. EdU incorporation assay was performed as we previously reported [56]. Briefly, cells were culture in 96-well plates at a density of 1000 cells per well. After 24 h, 50 μM EdU was added to each well and incubated for additional 2 h. The cells were fixed with 4% formaldehyde for 15 min and treated with 0.5% Triton X-100 for 20 min. After washing with PBS, 100 μl of 1 X Apollo reaction cocktail was added and incubated for 30 min. After staining with 100 μl of Hoechst 33342 for 30 min, the cells were visualized using EVOS cell image system. The results were analyzed by Image-Pro Plus 6.0 software (Media Cybernetics, USA). For the cell viability assay, 1 × 104 ATC cells expressed with the indicated constructs were seeded into 96-well microplates, doxorubicin was added after cell adhesion and cell viability was detected 24 h later as described in the CCK8 assay.
Cell migration analysis
For wound healing assay, cells were seeded into 6-well plates. When cells reached full confluence, a scratch was made on the cell layer with a 200 μl sterile pipette tip and washed with PBS. Cells were maintained in the medium containing 1% FBS and wound distance was measured every 24 h. Transwell migration assay was performed using 8 μm pore polycarbonate membrane transwell plates (Corning, USA). Briefly, 5 × 105 cells were suspended without serum and were seeded into the upper chambers of the transwell plates. The bottom chambers were filled with 600 μl complete medium. After 24 h, the migratory cells were fixed and stained with crystal violet.
ALDH activity assay
ALDH activity was measured using ALDH Activity Assay Kit (ab155893, Abcam) following the manufacturer’s instructions: NADH standard and sample were added into a 96-well plate in duplicate. Then the Reaction mix, containing Acetaldehyde and ALDH substrate, were added to each well to a final volume of 50 µl. Finally, the mixture was incubated for 1 h at room temperature and measured at OD 450 nm.
Sphere formation assay
In total, 2 × 103 single cells were seeded into 6-well ultra-low attachment culture plates (Corning, USA) in serum-free DMEM/F12 supplemented with B27 (1:50), 20 ng/ml EGF, and 20 ng/ml bFGF. Two weeks later, the spheres were photographed and counted.
In vivo tumorigenesis and metastasis assay
BALB/c nude mice aged 4 weeks were obtained from Beijing HFK Bioscience Co., Ltd. (Beijing, China). In total, 1 × 106 CAL-62 cells were injected to each mouse. Tumor sizes were measured every 3 days until the end of the experiment. For in vivo metastasis assays, 0.5 × 106 CAL-62 cells were intravenously injected into each mouse through the tail vein. The lungs were harvested 4 weeks after injection. The mice were maintained in a temperature and humidity‐controlled and specific pathogen‐free environment in the laboratory animal facility of Xiangya Hospital of Central South University. Tumor size was measured every 3 days until the end of the experiment. The experiments were performed under the protocols approved by ethnic committee of Xiangya Hospital of Central South University.
Limiting dilution assay in vivo
For the in vivo limiting dilution assay, spheroids were dissociated into single cells, serially diluted to the desired doses (5 × 103, 1 × 104, 5 × 104 and 1 × 105), and then subcutaneously injected into NOD-SCID mice. After 2 months, the number of tumors was counted, and the frequency of CSCs was assessed using ELDA software (http://bioinf.wehi.edu.au/software/elda/index.html) provided by the Walter and Eliza Hall Institute.
Statistical analysis
Student’s t test and one-way ANOVA were used to compare two and more groups respectively. Multiple comparison with—was performed when appropriate. A p value < 0.05 was considered as statistically significant and all tests were two-tailed. All statistical tests were performed with Prism 7.0 (GraphPad, USA).
References
Maniakas A, Dadu R, Busaidy NL, Wang JR, Ferrarotto R, Lu C, et al. Evaluation of overall survival in patients with anaplastic thyroid carcinoma, 2000-2019. JAMA Oncol. 2020;6:1397–404.
Capdevila J, Wirth LJ, Ernst T, Ponce Aix S, Lin CC, Ramlau R, et al. PD-1 blockade in anaplastic thyroid carcinoma. J Clin Oncol. 2020;38:2620–7.
Tiedje V, Stuschke M, Weber F, Dralle H, Moss L, Führer D. Anaplastic thyroid carcinoma: review of treatment protocols. Endocr Relat Cancer. 2018;25:R153–r61.
Kebebew E, Greenspan FS, Clark OH, Woeber KA, McMillan A. Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer. 2005;103:1330–5.
Molinaro E, Romei C, Biagini A, Sabini E, Agate L, Mazzeo S, et al. Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat Rev Endocrinol. 2017;13:644–60.
Smallridge RC, Ain KB, Asa SL, Bible KC, Brierley JD, Burman KD, et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104–39.
Xu B, Fuchs T, Dogan S, Landa I, Katabi N, Fagin JA, et al. Dissecting anaplastic thyroid carcinoma: a comprehensive clinical, histologic, immunophenotypic, and molecular study of 360 cases. Thyroid. 2020;30:1505–17.
Sasanakietkul T, Murtha TD, Javid M, Korah R, Carling T. Epigenetic modifications in poorly differentiated and anaplastic thyroid cancer. Mol Cell Endocrinol. 2018;469:23–37.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28.
Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014;15:642–56.
Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211–26.
Nguyen CDK, Yi C. YAP/TAZ signaling and resistance to cancer therapy. Trends Cancer. 2019;5:283–96.
Pobbati AV, Hong W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics. 2020;10:3622–35.
Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68:2592–8.
Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–72.
Mao W, Mai J, Peng H, Wan J, Sun T. YAP in pancreatic cancer: oncogenic role and therapeutic strategy. Theranostics. 2021;11:1753–62.
Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29:1271–84.
Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, et al. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer. 2014;13:110.
Zhou X, Li Y, Wang W, Wang S, Hou J, Zhang A, et al. Regulation of Hippo/YAP signaling and Esophageal Squamous Carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics. 2020;10:9443–57.
Wang Z, Kong Q, Su P, Duan M, Xue M, Li X, et al. Regulation of Hippo signaling and triple negative breast cancer progression by an ubiquitin ligase RNF187. Oncogenesis. 2020;9:36.
Zhang A, Wang W, Chen Z, Pang D, Zhou X, Lu K, et al. SHARPIN inhibits esophageal squamous cell carcinoma progression by modulating Hippo signaling. Neoplasia. 2020;22:76–85.
Li L, Liu T, Li Y, Wu C, Luo K, Yin Y, et al. The deubiquitinase USP9X promotes tumor cell survival and confers chemoresistance through YAP1 stabilization. Oncogene. 2018;37:2422–31.
Pan B, Yang Y, Li J, Wang Y, Fang C, Yu F, et al. USP47-mediated deubiquitination and stabilization of YAP contributes to the progression of colorectal cancer. Protein Cell. 2020;11:138–43.
Zhu H, Yan F, Yuan T, Qian M, Zhou T, Dai X, et al. USP10 promotes proliferation of hepatocellular carcinoma by deubiquitinating and stabilizing YAP/TAZ. Cancer Res. 2020;80:2204–16.
Farshi P, Deshmukh RR, Nwankwo JO, Arkwright RT, Cvek B, Liu J, et al. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat. 2015;25:1191–208.
Tang J, Tian Z, Liao X, Wu G. SOX13/TRIM11/YAP axis promotes the proliferation, migration and chemoresistance of anaplastic thyroid cancer. Int J Biol Sci. 2021;17:417–29.
Ciamporcero E, Daga M, Pizzimenti S, Roetto A, Dianzani C, Compagnone A, et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic Biol Med. 2018;115:447–57.
Huang C, Yuan W, Lai C, Zhong S, Yang C, Wang R, et al. EphA2-to-YAP pathway drives gastric cancer growth and therapy resistance. Int J Cancer. 2020;146:1937–49.
Valero V 3rd, Pawlik TM, Anders RA. Emerging role of Hpo signaling and YAP in hepatocellular carcinoma. J Hepatocell Carcinoma. 2015;2:69–78.
Zhang Z, Qiu N, Yin J, Zhang J, Liu H, Guo W, et al. SRGN crosstalks with YAP to maintain chemoresistance and stemness in breast cancer cells by modulating HDAC2 expression. Theranostics. 2020;10:4290–307.
Buckarma EH, Werneburg NW, Conboy CB, Kabashima A, O’Brien DR, Wang C, et al. The YAP-interacting phosphatase SHP2 can regulate transcriptional coactivity and modulate sensitivity to chemotherapy in cholangiocarcinoma. Mol Cancer Res. 2020;18:1574–88.
Tocci P, Cianfrocca R, Sestito R, Rosanò L, Di Castro V, Blandino G, et al. Endothelin-1 axis fosters YAP-induced chemotherapy escape in ovarian cancer. Cancer Lett. 2020;492:84–95.
Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–510.
Piersma B, de Rond S, Werker PM, Boo S, Hinz B, van Beuge MM, et al. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. 2015;185:3326–37.
Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell. 2014;26:273–87.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5.
Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6:27–37.
Crawford JJ, Bronner SM, Zbieg JR. Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review. Expert Opin Ther Pat. 2018;28:867–73.
Zhou Z, Zhou H, Ponzoni L, Luo A, Zhu R, He M, et al. EIF3H orchestrates Hippo pathway-mediated oncogenesis via catalytic control of YAP stability. Cancer Res. 2020;80:2550–63.
Yan C, Yang H, Su P, Li X, Li Z, Wang D, et al. OTUB1 suppresses Hippo signaling via modulating YAP protein in gastric cancer. Oncogene. 2022;41:5186–98.
Zhang Z, Du J, Wang S, Shao L, Jin K, Li F, et al. OTUB2 promotes cancer metastasis via Hippo-independent activation of YAP and TAZ. Mol Cell. 2019;73:7–21.e7.
Luo Y, Zhou J, Tang J, Zhou F, He Z, Liu T, et al. MINDY1 promotes bladder cancer progression by stabilizing YAP. Cancer Cell Int. 2021;21:395.
Fang Y, Fu D, Shen XZ. The potential role of ubiquitin c-terminal hydrolases in oncogenesis. Biochim Biophys Acta. 2010;1806:1–6.
Mtango NR, Sutovsky M, Susor A, Zhong Z, Latham KE, Sutovsky P. Essential role of maternal UCHL1 and UCHL3 in fertilization and preimplantation embryo development. J Cell Physiol. 2012;227:1592–603.
Suzuki M, Setsuie R, Wada K. Ubiquitin carboxyl-terminal hydrolase l3 promotes insulin signaling and adipogenesis. Endocrinology. 2009;150:5230–9.
Mtango NR, Sutovsky M, Vandevoort CA, Latham KE, Sutovsky P. Essential role of ubiquitin C-terminal hydrolases UCHL1 and UCHL3 in mammalian oocyte maturation. J Cell Physiol. 2012;227:2022–9.
Nishi R, Wijnhoven PWG, Kimura Y, Matsui M, Konietzny R, Wu Q, et al. The deubiquitylating enzyme UCHL3 regulates Ku80 retention at sites of DNA damage. Sci Rep. 2018;8:17891.
Zhang X, Smits AH, van Tilburg GB, Jansen PW, Makowski MM, Ovaa H, et al. An interaction landscape of ubiquitin signaling. Mol Cell. 2017;65:941–55.e8.
Kim JY, Lee JM, Cho JY. Ubiquitin C-terminal hydrolase-L3 regulates Smad1 ubiquitination and osteoblast differentiation. FEBS Lett. 2011;585:1121–6.
Song HM, Lee JE, Kim JH. Ubiquitin C-terminal hydrolase-L3 regulates EMT process and cancer metastasis in prostate cell lines. Biochem Biophys Res Commun. 2014;452:722–7.
Zhang MH, Zhang HH, Du XH, Gao J, Li C, Shi HR, et al. UCHL3 promotes ovarian cancer progression by stabilizing TRAF2 to activate the NF-kappaB pathway. Oncogene. 2020;39:322–33.
Song Z, Li J, Zhang L, Deng J, Fang Z, Xiang X, et al. UCHL3 promotes pancreatic cancer progression and chemo-resistance through FOXM1 stabilization. Am J Cancer Res. 2019;9:1970–81.
Ouyang L, Yan B, Liu Y, Mao C, Wang M, Liu N, et al. The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor. Signal Transduct Target Ther. 2020;5:78.
Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102.
Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–65.
Kong D, Li A, Liu Y, Cui Q, Wang K, Zhang D, et al. SIX1 activates STAT3 signaling to promote the proliferation of thyroid carcinoma via EYA1. Front Oncol. 2019;9:1450.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [82072594, YT; 82073097, 81874139, SL; 82002916, CM; 82073136, 81772927, DX; 82203160, JT], the China Postdoctoral Science Foundation [2019M652804, CM; 2022M713531, JT], the Natural Science Foundation of Hunan Province [2022JJ40815, JT; 2020JJ5790, CM], the Hunan Provincial Key Area R&D Programs [2019SK2253, YT], the Science and Technology Innovation Program of Hunan Province [2022RC3072 (YT)], and the Central South University Research Program of Advanced Interdisciplinary Studies (2023QYJC030, YT).
Author information
Authors and Affiliations
Contributions
GW and YT designed and supervised the research. JT, QY, CM, LZ, DX, SL, GW and YT performed research and provided helpful discussions. JT, QY, CM, LZ, DX, SL, GW and YT analyzed and interpreted the data. DX conducted pathology evaluations. All authors reviewed and edited the manuscript. KH contributed to all aspects of the study. QY performed animal model experiments. JT had a primary role in interpreting and organizing the data as well as writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The research was carried out according to the World Medical Association Declaration of Helsinki and was approved by the Ethics Committee at Xiangya Hospital of Central South University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Tang, J., Yang, Q., Mao, C. et al. The deubiquitinating enzyme UCHL3 promotes anaplastic thyroid cancer progression and metastasis through Hippo signaling pathway. Cell Death Differ 30, 1247–1259 (2023). https://doi.org/10.1038/s41418-023-01134-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-023-01134-z
- Springer Nature Limited
This article is cited by
-
GSK3β and UCHL3 govern RIPK4 homeostasis via deubiquitination to enhance tumor metastasis in ovarian cancer
Oncogene (2024)
-
The deubiquitinase EIF3H promotes hepatocellular carcinoma progression by stabilizing OGT and inhibiting ferroptosis
Cell Communication and Signaling (2023)