

Abstract
Arrhythmogenic cardiomyopathy (AC) is a clinically and genetically heterogeneous disorder of heart muscle that is associated with ventricular arrhythmias and risk of sudden cardiac death, particularly in the young and athletes. Mutations in five genes that encode major components of the desmosomes, namely junction plakoglobin, desmoplakin, plakophilin-2, desmoglein-2, and desmocollin-2, have been identified in approximately half of affected probands. AC is, therefore, commonly considered a 'desmosomal' disease. No single test is sufficiently specific to establish a diagnosis of AC. The diagnostic criteria for AC were revised in 2010 to improve sensitivity, but maintain specificity. Quantitative parameters were introduced and identification of a pathogenic mutation in a first-degree relative has become a major diagnostic criterion. Caution in the interpretation of screening results is highly recommended because a 'pathogenic' mutation is difficult to define. Experimental data confirm that this genetically determined cardiomyopathy develops after birth because of progressive myocardial dystrophy, and is initiated by cardiomyocyte necrosis; cellular and animal models are necessary to gain insight into the cascade of underlying molecular events. Crosstalk from the desmosome to the nucleus, gap junctions, and ion channels is under investigation, to move from symptomatic to targeted therapy, with the ultimate aim to stop disease onset and progression.
Key Points
-
Arrhythmogenic cardiomyopathy (AC) is a familial heart-muscle disease that is usually inherited with an autosomal-dominant pattern; mutations in desmosomal-protein genes are found in approximately 50% of probands
-
The 1994 diagnostic criteria were updated in 2010 to increase their sensitivity, but maintain their specificity; differential diagnosis with AC 'phenocopies' is mandatory when dealing with sporadic forms of AC
-
Emerging tools offer the possibility to visualize the fibrofatty scar, as either low-voltage myocardial areas using electroanatomical mapping, or areas of delayed contrast-enhancement with cardiac MRI
-
Genotype–phenotype studies show that the clinicomorphological spectrum of AC is wider than originally thought, and includes variants with predominant or even isolated left ventricular involvement within a single family
-
Animal and cellular models indicate that both abnormal biomechanical properties and crosstalk from the desmosome to the nucleus, gap junctions, and ion channels are implicated in the pathobiology of AC
-
Electrical instability is the main clinical manifestation of AC; in addition to re-entry arrhythmias caused by fibrofatty replacement, current hypotheses implicate acute cell death, gap-junction remodeling, and ion-channel crosstalk


Similar content being viewed by others

References
Online Medical Inheritance in Man®. 107970 Arrhythmogenic right ventricular dysplasia, familial, 1; ARVD1 [online], (2009).
Basso, C., Corrado, D., Marcus, F. I., Nava, A. & Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 373, 1289–1300 (2009).
Thiene, G., Nava, A., Corrado, D., Rossi, L. & Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 318, 129–133 (1988).
Corrado, D., Thiene, G., Nava, A., Pennelli, N. & Rossi, L. Sudden death in young competitive athletes: clinico-pathologic correlations in 22 cases. Am. J. Med. 89, 588–596 (1990).
Awad, M. M., Calkins, H. & Judge D. P. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, 258–267 (2008).
Basso, C. et al. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy or myocarditis? Circulation 94, 983–991 (1996).
Basso, C., Corrado, D. & Thiene, G. Arrhythmogenic right ventricular cardiomyopathy: what's in a name? From a congenital defect (dysplasia) to a genetically determined cardiomyopathy (dystrophy). Am. J. Cardiol. 106, 275–277 (2010).
Frank, R. et al. Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia. Arch. Mal. Coeur Vaiss. 71, 963–972 (1978).
Marcus, F. et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 65, 384–398 (1982).
Uhl, H. S. A previously undescribed congenital malformation of the heart: almost total absence of the myocardium of the right ventricle. Bull. Johns Hopkins Hosp. 91, 197–209 (1952).
McKenna, W. J. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 71, 215–218 (1994).
Richardson, P. et al. Report of the World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 93, 841–842 (1996).
Gallo P, d'Amati, G. & Pelliccia, F. Pathologic evidence of extensive left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy. Hum. Pathol. 23, 948–952 (1992).
Michalodimitrakis, M., Papadomanolakis, A., Stiakakis, J. & Kanaki, K. Left side right ventricular cardiomyopathy. Med. Sci. Law 42, 313–317 (2002).
Nava, A., Rossi, L. & Thiene, G. (Eds) Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (Elsevier, Amsterdam, 1997).
Marcus, F., Nava, A. & Thiene, G. (Eds) Arrhythmogenic RV Cardiomyopathy/Dysplasia: Recent Advances (Springer–Verlag, Milan, 2007).
Corrado, D., Basso, C., Thiene, G. Arrhythmogenic Cardiomyopathy, An Issue of Cardiac Electrophysiology Clinics (Elsevier, New York, 2011).
Corrado, D., Basso, C., Pilichou, K. & Thiene, G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 97, 530–539 (2011).
Marcus, F. I. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur. Heart J. 31, 806–814 (2010).
Asimaki, A. et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 360, 1075–1084 (2009).
Asimaki, A. et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 4, 743–752 (2011).
Tandri, H. et al. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J. Am. Coll. Cardiol. 45, 98–103 (2005).
Sen-Chowdhry, S. et al. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J. Am. Coll. Cardiol. 48, 2132–2140 (2006).
Sen-Chowdhry, S. et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J. Am. Coll. Cardiol. 52, 2175–2187 (2008).
Sen-Chowdhry, S. et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 115, 1710–1720 (2007).
Corrado, D. et al. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 111, 3042–3050 (2005).
Corrado, D. et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J. Am. Coll. Cardiol. 51, 731–739 (2008).
Garcia, F. C., Bazan, V., Zado, E. S., Ren, J. F. & Marchlinski, F. E. Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 120, 366–375 (2009).
Bauce, B. et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 26, 1666–1675 (2005).
Corrado, D. et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J. Am. Coll. Cardiol. 30, 1512–1520 (1997).
Nava, A. et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J. Am. Coll. Cardiol. 12, 1222–1228 (1988).
McKoy, G. et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355, 2119–2124 (2000).
Rampazzo, A. et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum. Mol. Genet. 3, 959–962 (1994).
Norgett, E. E. et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 9, 2761–2766 (2000).
Protonotarios, N. & Tsatsopoulou, A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 13, 185–194 (2004).
Kaplan, S. R. et al. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 13, 26–32 (2004).
Rampazzo, A. et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 71, 1200–1206 (2002).
Nava, A. et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 36, 2226–2233 (2000).
Gerull, B. et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 36, 1162–1164 (2004).
Pilichou, K. et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 1171–1179 (2006).
Syrris, P. et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 79, 978–984 (2006).
Asimaki, A. et al. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 81, 964–973 (2007).
Awad, M. M. et al. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 27, 1157 (2006).
Simpson, M. A. et al. Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology 113, 28–34 (2009).
Tiso, N. et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 10, 189–194 (2001).
Beffagna, G. et al. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 65, 366–373 (2005).
Merner, N. D. et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 82, 1–13 (2008).
van Tintelen, J. P. et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 6, 1574–1583 (2009).
Taylor, M. et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 124, 876–885 (2011).
Protonotarios, N. et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur. Heart J. 32, 1097–1104 (2011).
Quarta, G. et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 123, 1201–1209 (2011).
den Haan, A. D. et al. Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Cardiovasc. Genet. 2, 428–435 (2009).
Cox, M. G. et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype–phenotype follow-up study. Circulation 123, 2690–2700 (2011).
Bauce, B. et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 7, 22–29 (2010).
Xu, T. et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 55, 587–589 (2010).
Kapplinger, J. D. et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 57, 2317–2327 (2011).
Charron, P. et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 31, 2715–2726 (2010).
Delmar, M. & McKenna, W. J. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ. Res. 107, 700–714 (2010).
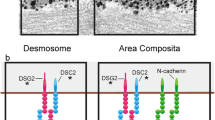
Franke, W. W., Borrmann, C. M., Grund, C. & Pieperhoff, S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur. J. Cell. Biol. 85, 69–82 (2006).
Mallat, Z. et al. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N. Engl. J. Med. 335, 1190–1196 (1996).
Valente, M. et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am. J. Pathol. 152, 479–484 (1998).
Pilichou, K. et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 206, 1787–1802 (2009).
Li, J. et al. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of β-catenin signaling. Mol. Cell. Biol. 31, 1134–1144 (2011).
Yang, Z. et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 99, 646–655 (2006).
Basso, C. et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur. Heart J. 27, 1847–1854 (2006).
Garcia-Gras, E. et al. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 116, 2012–2021 (2006).
Li, D. et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 20, 4582–4596 (2011).
Krusche, C. A. et al. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res. Cardiol. 106, 617–633 (2011).
Kaplan, S. R. et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 1, 3–11 (2004).
Sato, P. Y. et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526 (2009).
Sato, P. Y. et al. Interactions between ankyrin-G., plakophilin-2, and connexin 43 at the cardiac intercalated disc. Circ. Res. 109, 193–201 (2011).
Thiene, G. et al. Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? Eur. Heart J. 12, 22–25 (1991).
Calabrese, F., Basso, C., Carturan, E., Valente, M. & Thiene, G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: is there a role for viruses? Cardiovasc. Pathol. 15, 11–17 (2006).
Bowles, N. E., Ni, J., Marcus, F. & Towbin, J. A. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 39, 892–895 (2002).
Bonny, A. et al. C-reactive protein in arrhythmogenic right ventricular dysplasia/cardiomyopathy and relationship with ventricular tachycardia. Cardiol. Res. Pract. 2010, 919783 (2010).
Campian, M. E. et al. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 37, 2079–2085 (2010).
d'Amati, G., di Gioia, C. R., Giordano, C. & Gallo, P. Myocyte transdifferentiation: a possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Arch. Pathol. Lab. Med. 124, 287–290 (2000).
Lombardi, R. et al. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 104, 1076–1084 (2009).
Gittenberger-de Groot, A. C., Winter, E. M. & Poelmann, R. E. Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 14, 1056–1060 (2010).
Liang, W. C. et al. TMEM43 mutations in Emery–Dreifuss muscular dystrophy-related myopathy. Ann. Neurol. 69, 1005–1013 (2011).
Sen-Chowdhry, S. et al. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 3, 323–330 (2010).
Bauce, B. et al. Comparison of clinical features of arrhythmogenic right ventricular cardiomyopathy in men versus women. Am. J. Cardiol. 102, 1252–1257 (2008).
Pelzer, T. et al. 17β-Estradiol prevents programmed cell death in cardiac myocytes. Biochem. Biophys. Res. Commun. 268, 192–200 (2000).
Wlodarska, E. K. et al. Arrhythmogenic right ventricular cardiomyopathy in two pairs of monozygotic twins. Int. J. Cardiol. 105, 126–133 (2005).
Gonzalez-Juanatey, C., Testa, A. & Armesto, V. Identical twins with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Clin. Cardiol. 30, 529–530 (2007).
Pilichou, K., Bezzina, C. R., Thiene, G. & Basso, C. Arrhythmogenic cardiomyopathy: transgenic animal models provide novel insights into disease pathobiology. Circ. Cardiovasc. Genet. 4, 318–326 (2011).
Kirchhof, P. et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 114, 1799–1806 (2006).
Corrado, D., Basso, C., Rizzoli, G., Schiavon, M. & Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 42, 1959–1963 (2003).
Fabritz, L. et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J. Am. Coll. Cardiol. 57, 740–750 (2011).
Corrado, D. et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA 296, 1593–1601 (2006).
Pelliccia, A. et al. Outcomes in athletes with marked ECG repolarization abnormalities. N. Engl. J. Med. 358, 152–161 (2008).
Wichter, T., Borggrefe, M., Haverkamp, W., Chen, X. & Breithardt, G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 86, 29–37 (1992).
Wichter, T. et al. Arrhythmogenic right ventricular cardiomyopathy. Antiarrhythmic drugs, catheter ablation, or ICD? Herz 30, 91–101 (2005).
Marchlinski, F. E. et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation 110, 2293–2298 (2004).
Dalal, D. et al. Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 50, 432–440 (2007).
Corrado, D. et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 108, 3084–3091 (2003).
Corrado, D. et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 122, 1144–1152 (2010).
Bushby, K., Lochmüller, H., Lynn, S. & Straub, V. Interventions for muscular dystrophy: molecular medicines entering the clinic. Lancet 374, 1849–1856 (2009).
Acknowledgements
The authors are supported by Telethon, Rome; CARIPARO Foundation, Padova; and Veneto Region, Venice, Italy.
Author information
Authors and Affiliations
Contributions
C. Basso researched the data, and all the authors contributed substantially to discussion of the content of the article. C. Basso wrote the manuscript, and all the authors were involved in reviewing and editing it before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Basso, C., Bauce, B., Corrado, D. et al. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 9, 223–233 (2012). https://doi.org/10.1038/nrcardio.2011.173
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2011.173
- Springer Nature Limited
This article is cited by
-
Ringlike late gadolinium enhancement provides incremental prognostic value in non-classical arrhythmogenic cardiomyopathy
Journal of Cardiovascular Magnetic Resonance (2023)
-
Arrhythmogenic left ventricular cardiomyopathy caused by a novel likely pathogenic DSP mutation, p.K1165Rfs*8, in a family with sudden cardiac death
BMC Medical Genomics (2023)
-
A novel DSP zebrafish model reveals training- and drug-induced modulation of arrhythmogenic cardiomyopathy phenotypes
Cell Death Discovery (2023)
-
Left ventricular fibro-fatty replacement in arrhythmogenic right ventricular dysplasia/cardiomyopathy: prevalence, patterns, and association with arrhythmias
Journal of Cardiovascular Magnetic Resonance (2021)
-
Cardiovascular Magnetic Resonance Imaging and Heart Failure
Current Cardiology Reports (2021)