
Abstract
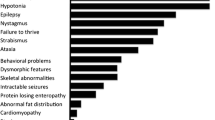
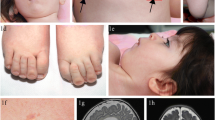
Summary: CDG Ie is caused by a deficiency of dolichol-phosphate-mannose synthase 1 (DPM1), an enzyme involved in N-glycan assembly in the endoplasmic reticulum. Three proteins are known to be part of the synthase complex: DPM1, DPM2 and DPM3. Only mutations in DPM1, the catalytic subunit, have been described in three families. One was homozygous for the c274C>;G (R92G) mutation in DPM1 and two others were compound heterozygous for R92G and a c628delC deletion or a c331–343del13, respectively. Clinical features were a severe infantile encephalopathy, early intractable seizures, acquired microcephaly, and some dysmorphic features. We report a patient with milder symptoms: microcephaly, dysmorphic features, developmental delay, optic atrophy, and cerebellar dysfunction without cerebellar atrophy. The patient is homozygous for a new mutation in exon 9 of the DPM1 gene (c742T>;C (S248P)). Our findings extend the spectrum of CDG Ie.
Similar content being viewed by others

REFERENCES
Briones P, Vilaseca MA, Garci´a-Silva MT, et al (2001) Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur J Paediatr Neurol 5: 127–131.
Burda P, Aebi M(1999 ) The dolichol pathway of N-linked glycosylation. Biochim Biophys Acta 1426: 239–257.
Colomé C, Ferrer I, Artuch R, Vilaseca A, Pineda M, Briones P (2000) Personal experience with the application of CDT assays to the detection of congenital disorders of glycosylation.Clin Chem Lab Med 38: 965–969.
Grünewald S, Matthijs G, Jaeken J (2002) Congenital disorders of glycosylation: A review. Pediatr Res 52: 618–624.
Imbach T, Schenk B, Schollen E, et al (2000) Defiiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J Clin Invest 105: 233–239.
Kim S, Westphal V, Srikrishna G, et al (2000) Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation type Ie (CDG-Ie). J Clin Invest 105: 191–198.
Kinoshita T, Satoshi T, Jun H, Kenji K, Yusuke M( 1999) Human dolichol phosphate mannose synthase consists of three subunits DPM1, DPM2 and DPM3. Glycobiology 9: 1104 [Abstract].
Schollen E, Martens K, Geuzens E, Matthijs G (2002) D-HPLC analysis as a platform for molecular diagnosis of congenital disorders of glycosylation (CDG). Eur J Hum Genet 10: 643–648.
Author information
Authors and Affiliations
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s10545-005-5824-6.
Rights and permissions
About this article
Cite this article
García-Silva, M.T., Matthijs, G., Schollen, E. et al. Congenital disorder of glycosylation (CDG) type Ie. A new patient. J Inherit Metab Dis 27, 591–600 (2004). https://doi.org/10.1023/B:BOLI.0000042984.42433.d8
Issue Date:
DOI: https://doi.org/10.1023/B:BOLI.0000042984.42433.d8