
Abstract
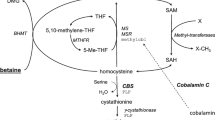
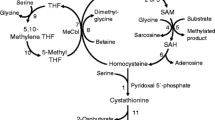
Summary: To explore the pathogenesis of cystathionine β-synthase (CBS) deficiency and to test the efficacy of pharmacological therapy we examined a panel of metabolites in nine homocystinuric patients under treated and/or untreated conditions. Off pharmacological treatment, the biochemical phenotype was characterized by accumulation of plasma total homocysteine (median 135 µmol/L) and blood S-adenosylhomocysteine (median 246 nmol/L), and by normal levels of guanidinoacetate and creatine. In addition, enhanced remethylation was demonstrated by low serine level (median 81 µmol/L), and by increased concentration of methionine (median 76 µmol/L) and N-methylglycine (median 6.8 µmol/L). Despite the substantially blocked transsulphuration, which was evidenced by undetectable cystathionine and severely decreased total cysteine levels (median 102 µmol/L), blood glutathione was surprisingly not depleted (median 1155 µmol/L). In 5 patients in whom pharmacological treatment was withdrawn, the differences of median plasma total homocysteine levels (125 µmol/L after withdrawal versus 33 µmol/L under treatment conditions), total cysteine levels (139 versus 211 µmol/L) and plasma serine levels (53 versus 103 µmol/L) on and off treatment demonstrated the efficacy of long-term pyridoxine/betaine administration (p<0.05). The treatment also decreased blood S-adenosylhomocysteine level (133 versus 59 nmol/L) with a borderline significance. In summary,our study shows that conventional treatment of CBS deficiency by diet and pyridoxine/betaine normalizes many but not all metabolic abnormalities associated with CBS deficiency. We propose that the finding of low plasma serine concentration in untreated CBS-deficient patients merits further exploration since supplementation with serine might be a novel and safe component of treatment of homocystinuria.
Similar content being viewed by others

REFERENCES
Allen RH, Stabler SP, Lindenbaum J (1993) Serum betaine, N,N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism 42: 1448–1460.
Bodamer OA, Bloesch SM, Gregg AR, Stockler-Ipsiroglu S, O'Brien WE (2001) Analysis of guanidinoacetate and creatine by isotope dilution electrospray tandem mass spectrometry. Clin Chim Acta 308: 173–178.
De Koning TJ, Duran M, Van Maldergem L, et al (2002) Congenital microcephaly and seizures due to 3-phosphoglyceraldehyde dehydrogenase deficiency: outcome of treatment with amino acid. J Inherit Metab Dis 25: 119–125.
Dudman NP, Tyrrell PA, Wilcken DE (1987) Homocysteinemia: depressed plasma serine levels. Metabolism 36: 198–201.
Helland S, Ueland PM (1983) Effect of 2'-deoxycoformycin infusion on S-adenosyl-homocysteine hydrolase and the amount of S-adenosylhomocysteine and related compounds in tissues of mice. Cancer Res 43: 4142–4147.
Janogik M, Oliveriusova J, Janogikova B, et al (2001) Impaired heme binding and aggregation of mutant cystathionine beta-synthase subunits in homocystinuria. Am J Hum Genet 68(6): 1506–1513.
Krijt J, Vackova M, Kojich V (2001) Measurement of homocysteine and other aminothiols in plasma: advantages of using TCEP as reductant compared to tri-n-butylphosphine. Clin Chem 47: 1821–1828.
Mudd HS, Poole JR (1975) Labile methyl balances for normal humans on various dietary regimens. Metabolism 24: 721–735.
Mudd SH, Levy HL, Kraus JP (2001) Disorders of transsulphuration. In Scriver CR, Beaudet AL, Sly WS, Valle D, eds; Childs B, Kinzler KW, Vogelstein B, assoc. eds. The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 2007–2056.
Stabler SP, Lindenbaum J, Savage DG, Allen RH (1993) Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood 81: 3404–3413.
Stabler SP, Sampson DA, Wang LP, Allen RH (1997) Elevations of serum cystathionine and total homocysteine in pyridoxine, folate and cobalamin deficient rats. Nutr Biochem 8: 279–289.
Stabler SP, Steegborn C, Wahl MC, et al (2002) Elevated plasma total homocysteine in severe methionine adenosyltransferase I/III deficiency. Metabolism 51(8): 981–988.
Togawa T, Sengupta S, Chen H, et al (2000) Mechanisms for the formation of protein-bound homocysteine in human plasma. Biochem Biophys Res Commun 277(3): 668–674.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Orendáč, M., Zeman, J., Stabler, S.P. et al. Homocystinuria due to cystathionine β-synthase deficiency: Novel biochemical findings and treatment efficacy. J Inherit Metab Dis 26, 761–773 (2003). https://doi.org/10.1023/B:BOLI.0000009963.88420.c2
Issue Date:
DOI: https://doi.org/10.1023/B:BOLI.0000009963.88420.c2