
Abstract
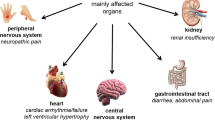
Recent clinical trials have demonstrated that enzyme replacement therapy with α-galactosidase A (α-Gal A) constitutes a major clinical advance in the treatment of patients with Fabry disease. This new therapeutic approach has been shown to be well tolerated and effective in reducing levels of the storage product globo-triaosylceramide and in normalizing many of the debilitating manifestations of the disorder. A double-blind placebo-controlled trial in 26 hemizygous male patients showed that agalsidase alfa (human α-Gal A) significantly reduced neuropathic pain (p= 0.02), increased creatinine clearance (p = 0.02), improved glomerular histology, reduced the QRS interval on electrocardiography and increased weight gain. Positron emission tomography also revealed normalization of cerebrovascular flow. After the 6-month controlled period, all patients were given agalsidase alfa for a further12 months. At the end of this period, all patients had a decrease in neuropathic pain, and there was a significant improvement in their ability to sense heat and cold. In addition, renal function stabilized, even in patients with renal insufficiency at the onset of treatment, and patients reported a normalization of sweating and improvements in their level of energy and sense of well-being. These findings show that enzyme replacement therapy offers promise as an effective management strategy for patients with Fabry disease.
Similar content being viewed by others


REFERENCES
Barton NW, Brady RO, Dambrosia JM, et al (1991) Replacement therapy for inherited enzyme deficiency-macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med 324: 1464–1470.
Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO (1990) Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci USA 87: 1913–1916.
Brady RO (1966) Sphingolipidoses. N Engl J Med 275: 312–318.
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med 276: 1163–1167.
Brady RO, Tallman JF, Johnson WG, et al (1973) Replacement therapy for inherited enzyme deficiency: use of purified ceramidetrihexosidase in Fabry's disease. N Engl J Med 289: 9–14.
Brady RO, Uhlendorf BW, Jacobson CB (1971) Fabry's disease: antenatal detection. Science 172: 174–175.
de Duve C (1964) From cytases to lysosomes. Fed Proc 23: 1045–1049.
Desnick RJ, Dean KJ, Grabowski G, Bishop DF, Sweeley CC (1979) Enzyme therapy in Fabry disease: differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. Proc Natl Acad Sci USA 76: 5326–5330.
Eng CM, Cochat P, Wilcox WR, et al. (2000) Enzyme replacement therapy in Fabry disease: Results of a placebo-controlled phase 3 trial. Am J Hum Genet 67(Suppl 2): 38.
Grabowski GA, Barton NW, Pastores G, et al (1995) Enzyme therapy in Gaucher disease Type 1: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Int Med 122: 33–39.
Johnson WG, Brady R (1972) Ceramidetrihexosidase from human placenta. Methods Enzymol XXVIII: 849–856.
Kusiak JW, Quirk JM, Brady RO (1978) Purification and properties of the two major isozymes of α-galactosidase from human placenta. J Biol Chem 253: 184–190.
Ohshima T, Murray GJ, Swain WD, et al (1997) α-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci USA 94: 2540–2544.
Schiffmann R, Kopp JB, Austin HA, et al. (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285: 2743–2749.
Schiffmann R, Murray GJ, Treco D, et al (2000) Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA 97: 365–370.
Weinreb NJ, Brady RO, Tappel AL (1968) The lysosomal localization of sphingolipid hydrolases. Biochim Biophys Acta 159: 141–146.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Brady, R.O., Murray, G.J., Moore, D.F. et al. Enzyme replacement therapy in Fabry disease. J Inherit Metab Dis 24 (Suppl 2), 18–24 (2001). https://doi.org/10.1023/A:1012451320105
Issue Date:
DOI: https://doi.org/10.1023/A:1012451320105