
Abstract
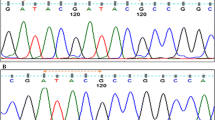
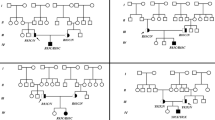
A novel mutation, C118t, in exon 2 of the acid α-glucosidase gene has been found in an infant with glycogen storage disease type II. This mutation is predicted to result in protein truncation. The phenotype was that of the severe infantile form of the disorder with lack of motor development, but with eye regard, social smile and vocalization. The parents were heterozygous for C118T and belong to an Islamic community opposed to termination of pregnancy. As the C118T mutation results in the loss of one of two AvaI sites present in an informative PCR product, reliable premarriage carrier detection became possible and was acceptable to the members of this extended family.
Similar content being viewed by others

REFERENCES
Hirschhorn R (1995) Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease, 7th edn. New York: McGraw-Hill, 2443–2464.
Huie ML, Chen AS, Sklower Brooks S, Grix A, Hirschhorn R (1994) A de novo 13nt deletion, a newly identified C647W missense mutation and a deletion of exon 18 in infantile onset glycogen storage disease type II (GSDII). Hum Mol Genet 3: 1081–1087.
Reuser AJJ, Kroos MA, Oude Elferink RP, Tager JM (1985) Defects in synthesis, phosphorylation, and maturation of acid α-glucosidase in glycogenosis type II. J Biol Chem 260: 8336–8341.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Kroos, M.A., Waitfield, A.E., Joosse, M. et al. A novel acid α-glucosidase mutation identified in a Pakistani family with glycogen storage disease type II. J Inherit Metab Dis 20, 556–558 (1997). https://doi.org/10.1023/A:1005394706622
Issue Date:
DOI: https://doi.org/10.1023/A:1005394706622