
Abstract
Anaerobic methanotrophic archaea (ANME) can assimilate methane and govern the greenhouse effect of deep-sea cold seeps. In this study, a total of 13 ANME draft genomes representing five ANME types (ANME-1a, ANME-1b, ANME-2a, ANME-2b and ANME-2c), in size between 0.8 and 1.8 Mbp, were obtained from the Jiaolong cold seep in the South China Sea. The small metagenome-assembled genomes (MAGs) contained all the essential pathways for methane oxidization and carbon dioxide fixation. All genes related to nitrate and sulfate reduction were absent from the MAGs, indicating their syntrophic dependence on partner organisms. Aside from acetate secretion and sugar storage, propanoate synthesis pathway, as an alternative novel carbon flow, was identified in all the MAGs and transcriptionally active. Regarding type-specific features of the MAGs, the genes encoding archaellum and bacteria-derived chemotaxis were specific to ANME-2, perhaps for fitness under fluctuation of methane and sulfate concentration flux. Our genomic and transcriptomic results strongly suggested that ANME could carry out simple carbon metabolism from C1 assimilation to C3 biosynthesis in the SCS cold seep, which casts light on a novel approach for synthetic biology.
Similar content being viewed by others

Introduction
The cold seeps on the global seafloors are the habitats of chemolithoautotrophic microorganisms that form a unique microbial community structure (Levin 2005). In these ecosystems, the anaerobic oxidation of methane (AOM) coupled with sulfate reduction is an important biogeochemical process in the subsurface sediments with methane leakage (Bowles and Joye 2011). The anaerobic methanotrophic archaea (ANME) are the key players in the methane assimilation with sulfate and nitrate as election acceptors (Boetius et al. 2000; Ettwig et al. 2010; Haroon et al. 2013; Scheller et al. 2016). In a cold seep of high methane flow, ANME spread rapidly and must compete for electron acceptors (Boetius and Wenzhofer 2013). ANME-1 and ANME-2 have syntrophic sulfate-reducing bacteria (SRB) that accept electrons from the ANME (Oni and Friedrich 2017). Energy metabolism of ANME has been elaborated by previous studies that focused on associations of cross-membrane electron transfer and methane oxidation process (Oni and Friedrich 2017). CO2, as the product of methane oxidation, might be further fixed into central carbon metabolism via Wood–Ljungdahl (W–L) pathway (Ragsdale and Pierce 2008). Alternatively, the reductive acetyl-CoA pathway was probably active in ANME-1 with genomics evidence (Cui et al. 2015). Although having an incomplete pathway, ANME was deduced to carry out gluconeogenesis (Skennerton et al. 2017). In ANME-2, acetate was regarded as a fermentation product of acetyl-CoA and could be secreted to the syntrophic SRB probably as carbon source. Understanding the carbon flow initiated from methane assimilation in ANME is critical for us to evaluate their role in cold seep ecosystem and global carbon budget. Now, bioinformatics pipelines may allow for binning of high-quality draft genomes of microbial inhabitants in a complicated assembly of different strains such as deep-sea marine sediments (Tyson et al. 2004). In cold seeps, different types of ANME always coexist (Wu et al. 2018), which renders difficulties in separating their genomes from a metagenome. Most of the draft genomes were obtained from enriched samples in bioreactors. Overall, due to a lack of high-quality genomes and transcriptomes from natural environments, the carbon metabolisms were still controversial.
The basement structure at the northern slope of the South China Sea (SCS) is complex with newly developed tectonic faults (Feng et al. 2015; Larsen et al. 2018), indicating that the sediments under the slope are favorable for gas hydrate accumulation and storage. Several methane seepage vents and reefs have been located and investigated recently in the northeastern part of the SCS (Cui et al. 2016; Shen et al. 2014; Zhang et al. 2012). The Jiaolong cold seep, which was discovered in 2013, had developed a typical cold-seep ecosystem. In this area, the overflow of methane could be clearly detected by geochemical surveys and an in situ Raman spectrometry measurements (Du et al. 2018b; Feng et al. 2015). In a recent study, we reported on the structure of microbial communities and the distribution of functional genes of the core players in four subsurface sediment layers of the Jiaolong cold seep (Wu et al. 2018). In this study, we obtained 13 ANME genomes representing two subtypes of ANME-1 and three subtypes of ANME-2 from the metagenomes of the Jiaolong cold seep. The compact genomes help understand a competitive relationship between the ANME types in the high-methane-flow sediments and intrinsic metabolic diversification of the different ANME types.
Results and discussion
Small ANME MAGs
Four sediment layers of the Jiaolong cold seep were used for metagenomics sequencing: S1: 0–2 cm below surface (cmbsf), S3: 4–6 cmbsf, S5: 8–10 cmbsf and S7: 12–14 cmbsf. The microbial communities and functional gene profiles of the four layers had been reported previously (Wu et al. 2018). Here, 45 Gbp Illumina 2 × 250 bp data for the four sediment layers of one core were obtained. After quality filtration, about 42.4 Gbp of clean data were used for assembly between neighboring layers and 13 ANME MAGs were binned. Their sizes were between 0.8 and 1.8 Mbp and GC content ranged between 43.3 and 52.5% (Table 1). Genome completeness was estimated to be 77.7–98.8% with a contamination rate of up to 5.9%. In contrast, the reference ANME genomes were all over 3 Mbp in size. The MAGs presented here were much smaller than other known ones (Wang et al. 2014). Note that the GC contents of ANME-2c MAGs were all higher than 50%, which indicates that amino acid composition and biological evolution rate of ANME-2c were different from other ANME types (Du et al. 2018a).
Phylogenetic and pangenomic analysis
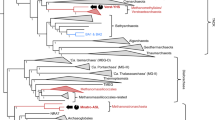
16S rRNA genes could be identified in eight of the ANME MAGs. A phylogenetic tree based on the 16S rRNA genes showed that the MAGs were derived from ANME-1, ANME-2a, -2b and ANME-2c (Fig. 1). The MAGs JLS104-2b, JLS301-2b, JLS503-2b and JLS702-2b were clustered with known ANME-2ab sequences and showed affinity to a cloning sequence from the same region. The short genetic distance between the sequences from the MAGs indicates that they were from the same species distributed in all four layers. This clade was independent of the neighboring one, consisting of those ANME-2ab from elsewhere, indicating the endemism of the ANME in the Jiaolong cold seep. The 16S rRNA genes from three MAGs JLS502-2c, JLS704-2c and JLS703-2c were grouped into two branches in ANME-2c, suggesting that there were at least two species in the ANME-2c in the core. The 16S rRNA gene from JLS501-1a MAG was similar to an ANME-1 16S rRNA gene from the Eel River Basin. In our previous work, ANME-1a archaea were present almost exclusively in S5 and S7 (Wu et al. 2018).

Maximum-likelihood phylogenetic tree of 16S rRNA genes. The 16S rRNA genes were extracted from ANME MAGs in this study and were marked in red. A total of 175 rRNA gene sequences for methanogens and ANME were pooled for the reconstruction of the tree
A phylogenomic tree based on 24 commonly conserved proteins from all the MAGs and several reference genomes displayed the relationships between the MAGs presented here and the genomes of known ANME and methanogens. The result showed that the MAGs presented here all belonged to ANME affiliated with 5 types (ANME-1a, ANME-1b, ANME-2a, ANME-2b and ANME-2c) (Fig. 2). JLS501-1a and JLS701-1a were clustered together and related to ANME-1a. Five MAGs were assigned to ANME-2c, in which two groups were formed as shown in the maximum-likelihood (ML) tree of the 16S rRNA genes. The two ANME-2b MAGs from S1 and S3 were also distantly related to the two from S5 and S7. This was consistent with the stratification of microbial communities across the layers (Wu et al. 2018), which was probably arose from the distribution of electron acceptors. SRB, as the associated electron acceptors of ANME, were restricted in S5 and S7 (Wu et al. 2018), which determined the distribution of ANME types that depend on the SRB syntrophic partner.

Genome features and gene contents of methanogens and ANME. The maximum-likelihood (ML) phylogenetic tree was constructed using 24 commonly conserved proteins from ANME MAGs and reference genomes. The bootstrap values were shown at the nodes of the tree with 1000 replicates. The MAGs from this study were noted with an asterisk. The genome features were illustrated by bubble charts. The heatmap shows COG annotation of the genomes. The COG numbers of individual COG categories were compared between reference genomes and MAGs by two-tailed t-test (*P < 0.5; **P < 0.01). The COG categories are described in COG database (https://www.ncbi.nlm.nih.gov/COG)
Clusters of orthologous groups (COGs) in the ANME and methanogen genomes were classified into functional categories (Fig. 2). Carbohydrate transport and metabolism (G), transcription (K), replication, recombination and repair (L), energy production and conversion (C), posttranslational modification (O), and inorganic ion transport and metabolism (P) were particularly enriched in the reference genomes, which ultimately resulted in their genome expansion. For most of the categories, the relationships between the COG numbers, the reference genomes and the MAGs reported here were significant (t-test; P < 0.01).
The ANME pangenomic analysis revealed a total of 5409 unique COG clusters that had been diversified among the genomes. Five clustering bins were specific to three reference ANME genomes (Fig. 3 and supplementary Table S1). Among them, there were 127 COGs for the ANME-2c type, and 205 for the ANME-1 type. Some of the COGs were annotated to the same function but sequence similarity was below the cutoff value (supplementary Table S1). The average nucleotide identity (ANI) analysis divided the ANME genomes into five types (Fig. 3), consistent with the results shown in the phylogenomic tree. In particular, ANME-2c MAGs were divided into two groups in the ANI result. In one of the groups, JLS703-2c and JLS502-2c possessed 385 specific COGs.

Pangenomic analysis of gene clusters within ANME MAGs. The COGs were compared and clustered among MAGs and reference ANME genomes (see Table 1 for details). The outmost circle exhibits the COGs in the genomes. The tiny brown bar denotes the COG cluster containing the single conserved genes (SCGs) shared by all the genomes. The Anvi’o also provided the number of singleton gene clusters that were present in only one of the genomes; the number of gene clusters; the number of genes per Kbp genomic region; an evaluation of genome completion. The numbers in the brackets following these items are the maximum values in the genomes. The COGs in the five selected subtype-specific bins (bin 1-5) were shown in supplementary Table S1
Metabolic pathway
The Jiaolong cold seep was active and the concentration of methane in sediments had been documented (0 cm: 0 mmol/L, 20 cm: 6.36 ± 0.11 mmol/L, 30 cm: 6.87 ± 0.33 mmol/L, 40 cm: 16.88 ± 0.33 mmol/L) in a previous study (Du et al. 2018b). The δ13C values of CH4 dissolved in seawater has also been measured (the δ13C values of two water samples: − 58.7‰ and − 61.1‰) (Feng et al. 2015). Over 70% of the methane discharged from the bottom to the sediment surface was consumed by microorganisms, and provided a carbon source and energy for the chemoautotrophs (Knittel et al. 2005). From the annotation results of the predicted proteins, and almost complete methane oxidation pathway in the ANME-2 MAGs and the reference genomes (Fig. 4) was obtained. Probably due to genome incompleteness, the ANME-1 MAGs JLS701-1a, JLS501-1b and JLS103-1b did not contain all the genes necessary for the AOM process (supplementary Table S2). As a cofactor of methyl group transfer in the AOM (Glass et al. 2014), vitamin B12 was synthesized by the ANME in the samples reported here, as the related genes were identified in the genomes. ANME-1 and ANME-2 used different types of electron transfer systems to drive the AOM. Both the reference genomes of ANME-2a and ANME-2d had a complete operon, consisting of fpoABCDHIJKLMNO gene cluster encoding the F420H2 dehydrogenase to participate the energy-conserving electron transport system (Baumer et al. 2000). The fpo operon was also present in JLS704-2c MAGs, only lacking fpoO. The JLS301-2b and JLS101-2a MAGs contained only fpoLMNO genes that appeared at the end of contigs. The truncation of fpo operon was likely a result of genome incompleteness. The reference ANME-1b genome had an Fqo system including fqoANLMKJBCDHI genes, which differed from the known gene arrangement in Archaeoglobus fulgidus (Bruggemann et al. 2000). There were only fqoLMK genes in the JLS501-1a and JLS103-1b MAGs and other genes were absent, likely also due to genome incompleteness. Previous studies had reported that ANME-1 type might contain the Fpo system but this seemed not to be the case in the ANME-1 MAGs reported here (McGlynn 2017). The AOM required the involvement of F420 as a cofactor. Dozens of radical S-adenosylmethionine genes and multiple copies of Fe-S oxidoreductase genes, most of which might take part in F420 synthesis, were identified here (Mehta et al. 2015).

Schematic metabolism of the ANME types. Type-specific metabolic pathways were depicted within the dashed box or labeled with a shaded name. The red arrows indicated the pathway of propanoate production. Abbreviations for enzymes and co-factors were shown in supplementary Table S4
From carbon dioxide to acetyl-CoA, the Wood-Ljungdahl (WL) pathway was used to carry out the assimilation. The related WL genes were all present in the MAGs. From acetyl-CoA to pyruvate, CO2 might also be fixed by pyruvate ferredoxin oxidoreductase (Por), since the genes encoding the Por complex were identified in the MAGs (supplementary Table S3). The ANME also potentially carried out other C1 metabolic processes. Formaldehyde could be catabolized into central carbon metabolism by integrating with ribulose and CH2=H4MPT (Fig. 4). Formate might also be utilized by first converting to CO2. The methanol dehydrogenase (mdo) gene that took part in conversion of formate and formaldehyde was present only in ANME-2 genomes.
From pyruvate, glucose might be generated as a storage product of organic matter, but some of the genes involved in gluconeogenesis were lacking in all the ANME genomes, as shown in previous studies (Skennerton et al. 2017). A propanoate synthesis pathway for all the ANME types via citraconate and 2-oxobutanoate (Fig. 4) was found in the MAGs. This propanoate-producing pathway has never previously been reported in the ANME and only rarely in other prokaryotes. Producing and releasing propanoate to the environment via diffusion was probably a mechanism to remove excess organic carbon derived from rapid methane assimilation. Furthermore, ANME-1 MAGs contained a butanoyl-CoA synthesis pathway. How the butanoyl-CoA further metabolized was remained unknown. None of the ANME MAGs and reference genomes had a complete set of genes for the citrate cycle (TCA cycle) (supplementary Table S3). In the bacterial production of propanoate, glucose was fermented to oxaloacetate, followed by CO2 fixation to generate propionyl-CoA and then propanoate (Liu et al. 2012). Such an approach for propanoate generation differed from that in ANME. Homologs which were similar to the propanoate-producing genes in the ANME MAGs were searched in NCBI. Results presented here showed that cimA, leuBCD and ACSS could be found in ANME and methanogenic archaea, including ANME-2, Methanococcoides methylutens, Methanosarcinales archaea, and Methanophagales archaea (supplementary Table S5). This suggested that the propionate synthesis pathway was also present in other archaeal genomes, but never been discerned.
Propanoate, as a common organic material, was mainly used as a food preservative, anti-bacterial agent, nitrocellulose solvent, plasticizer and chemical reagent (Liu et al. 2012). Derivatives of propanoate were also used to make perfumes, pesticides, and pharmaceuticals (Liu et al. 2012). Generally, microorganisms such as Propionibacterium acidipropionici and Propionibacterium shermanii could utilize a variety of fermentable sugars to produce propanoate (Gardner and Champagne 2005). Propanoate also had a variety of industrial synthesis methods (Schulz and Kluytmans 1983; Kumar and Babu 2006; Tyree et al. 1991). The propanoate synthesis pathway of ANME in this study might be another biosynthetic method to fill the demand for propanoate. However, the slow growth rate of ANME-2, due to it’s a syntrophic mode of life, precluded its industrial value as an efficient propanoate producer. The capacity of ANME-1 to be independent of a syntrophic partner provides perspectives for its application in transforming methane to propanoate.
suuACB genes were present in all ANME MAGs and genomes, indicating that the ANME could transport and utilize alkanesulfonate. Probably, sulfite, as a by-product of alkanesulfonate degradation, would be fed into cysteine synthesis pathway. In ANME-1b, sulfite might be combined with phosphoenolpyruvate, with the involvement of comABNED genes present in the MAGs, to synthesize sulfoacetaldehyde (Graupner et al. 2000). Except for NifH gene, that was involved in the conversion of nitrogen to ammonia, there were no more genes encoding nitrogen metabolism in the MAGs. In the ANME-1b MAG reported here, only the genes encoding nitrate ABC transporter and cytochrome C nitrite reductase small subunit (NrfH) were present. This was also true for the reference ANME-1b genome.
The ANME-2 MAGs reported here had a chemotaxis system encoded by the cheABCDWY genes and an archaellum-coding operon (flaBDEFHIJ). The archaellum in ANME-2 was a Euryarchaeota specific motility system coupled with the chemotaxis system obtained through HGT from bacteria (Albers and Jarrell 2018). The che genes were most similar to the homologs in Desulfuromonas sp. with a similarity between 31% (cheC in JLS101-2a) and 90% (cheY in JLS502-2c). The motility structure has not previously been reported by in an ANME-2 study (Wang et al. 2013) nor in ANME-1 genomes. With chemotaxis and motility, the ANME-2 archaea were probably more sensitive to sulfate gradients, due to the syntrophic association with SRB bacteria. Sensing and adapting to changes by the ANME were thus anticipated to play a major role in shaping microbial communities, in affecting dynamics of microbial activities, as well as in influencing various microbial responses to their surroundings (Miller et al. 2009). In the sediment core, the ANME-2 archaea were restricted to the S5 and S7 layers (Wu et al. 2018). The advantage of ANME-2 chemotaxis and movement might also enable the detection of methane flux and nutrients.
Transcriptomic evidence for propanoate producing activities
The in situ activity of the ANME types could be inferred from the transcriptomes of the four samples. The functional genes for methane fixation were abundant in the transcripts for the S3, S5 and S7 layers (Fig. 5). Nevertheless, fpoAK gene transcripts were absent from the S3 transcriptome, indicating decreasing methane assimilation at this layer (supplementary Fig. S1). In the S1 transcriptome, fpoAIKN, mdo, korA, leuC and pfk gene transcripts were not detected, which was consistent with an almost lack of methane oxidation therein (Wu et al. 2018). In the transcriptomes, the transcriptional levels of cimA, acs, and ACSS genes involved in propanoate generation, were also remarkably high at the S3–S7 sediment layers, which correlated with the transcriptional levels of methane-oxidizing genes in the corresponding layers. In particular, cimA as the first functional gene obligate for the step in pyruvate to citramalate in the propanoate synthesis pathway was abundantly transcribed in the three transcriptomes (Howell et al. 1999). Since Acs and ACSS were also involved in acetate production (Fujino et al. 2001), the transcriptomic results presented here suggested that acetate was also a product of methane oxidation.

Estimate of transcriptional levels of functional genes. The enzymes encoded by Acs and acsA (marked with *) genes can also catalyze acetyl coenzyme A to acetate. eno enolase, gpm putative phosphoglycerate mutase, pgk phosphoglycerate kinase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, fba fructose-bisphosphate aldolase, fbp bifunctional inositol-1 monophosphatase/fructose-1,6-bisphosphatase, pgi putative glucose-6-phosphate isomerase
Glucose synthesis was quite active, as indicated by the abundant gene transcripts for the gluconeogenesis/glycolysis pathway (Fig. 5). Except for at the S1 layer, the transcriptional level of cimA gene at the other layers was higher than that of ppdk (supplementary Fig. S1), indicating that pyruvate was prone to be apportioned to propionate rather than to glucose at the S3–S7 layers. The strong methane assimilation at S5 and S7 layers probably caused a high level of sugars in the ANME cells, which then raised the suppression of the glucose synthesis pathway to some extent. In this circumstance, acetate and propanoate as an alternative carbon flow were likely employed to deplete the extra carbon from the cells.
Since both propionate and acetate production could be mediated by ACSS (Fujino et al. 2001), it was unlikely to distinguish between and quantify propionate and acetate in the carbon low in this study. Biochemical and microbial physiological experiments using cultivated ANME strains would confirm the production of propanoate and/or acetate under optimized growth conditions.
Conclusions
In this study, we obtained draft genomes of ANME archaea and revealed their metabolic diversity in an active methane seepage site. In particular, we found a novel carbon flow that initiated from methane assimilation—the formation of propionic acid, aside from the predicted approaches leading to acetate and glucose in ANME-2. The chemotaxis system was only found in the ANME-2 type. The various types of ANME co-existed in the cold seep under environmental selection that probably encouraged microbial evolutionary differentiation for competitiveness in the cold-seep microenvironment. The methane biological assimilation efficiency that differed between the sediment layers with ANME inhabitants would be further explored using more MAGs and their transcriptomic profiles.
Materials and methods
Sampling and metagenomic sequencing
A sediment pushcore was collected at Jiaolong cold seep (22° 07′ N, 119° 17′ E) by the Jiaolong manned submersible in June 2013 at a depth of 1143 m; it was divided into four layers as described previously (Liu et al. 2014; Wu et al. 2018).
Genomic DNA was extracted from 2 g sediment from each layer using MoBio PowerSoil DNA isolation kit (Mo Bio, Carlsbad, CA, USA). About 200 ng genomic DNA was fragmented to ~ 550 bp by ultrasonication. Genomic libraries were built with TruSeq Nano DNA Library kit (Illumina, San Diego, CA, USA) and sequenced on a HiSeq2500 platform (Illumina, San Diego, CA, USA). After quality control and filtration of low-quality data, about 10.6 Gbp clean data of each layer were assembled using by SPAdes (v3.11) (Nurk et al. 2013).
Genome binning and annotation
The coverage of scaffolds was calculated by mapping the reads on the scaffolds with Bowtie 2 (Langmead and Salzberg 2012). As reported by Albertsen et al. (2013), the genome binning was performed using two-dimensional separation of the coverage levels in neighboring layers and correspondence analysis of tetra-nucleotide frequency (TNF) of the scaffolds (supplementary Fig. S2). The completeness and contamination rate of the MAGs were assessed by CheckM (v1.0.5) (Parks et al. 2015) using 85 conserved single-copy genes universally present in archaeal genomes (Wang et al. 2019).
CDSs and proteins in the MAGs were predicted by Prodigal (v2.6.2) (Hyatt et al. 2012). Annotation of CDSs was carried out using BLASTp against databases including NCBI non-redundant protein (NR) database, KEGG database (Kanehisa and Goto 2000), and COG database (Tatusov et al. 2000) with an e-value cutoff of 1e-05. Pseudogenes were identified by Prokka (Seemann 2014). The functional genes for AOM process were identified from the result of InterProScan (v5.28) with default settings (Quevillon et al. 2005; Zdobnov 2001).
Phylogenetic analysis
The 16S rRNA genes in the scaffolds were identified using rRNA_HMM (Huang et al. 2009). The commonly conserved proteins were identified using hmmsearch (3.0) (Krogh et al. 1994). The reference genomes and 16S rRNA genes were downloaded from the NCBI and Integrated Microbial Genomes (IMG) database. The 16S rRNA genes and 24 commonly conserved proteins (supplementary Table S6) were aligned with MAFFT E-INS-i (v7.294b) separately (Alva et al. 2016). The alignments were concatenated and adjusted with trimAl (v1.4.rev15) (Capellagutiérrez et al. 2009). To build the maximum-likelihood algorithm, the trees were inferred by ML algorithm using raxmlGUI v1.5 (Silvestro and Michalak 2012). 1000 replicates were performed to obtain bootstrap values.
Microbial pangenomics analysis
Microbial pangenomics analysis was performed using Anvi’o workflow (Delmont and Eren 2018; Eren et al. 2015). The MAGs and three reference genomes were converted into an Anvi’o contigs database. Subsequently, amino acid sequences were searched using BLASTp against COG database. The minbit for mcl clustering was set to 1. The pairwise ANI of the MAGs and the three reference ANME genomes was calculated using PyANI integrated in Anvi’o.
Data availability
The assembled genome bins have been deposited in the BIG Data Center (https://bigd.big.ac.cn) and can be accessed with BioProject number PRJCA000792. The genome accession numbers are GWHAAFW00000000, GWHAAFX00000000, GWHAAFY00000000, GWHAAFZ00000000, GWHAAGA00000000, GWHAAGB00000000, GWHAAGC00000000, GWHAAGD00000000, GWHAAGE00000000, GWHAAGF00000000, GWHAAGG00000000, GWHAAGH00000000 and GWHAAGI00000000.
References
Albers SV, Jarrell KF (2018) The archaellum: An update on the unique archaeal motility structure. Trends Microbiol 26:351–362
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538
Alva V, Nam SZ, Soding J, Lupas AN (2016) The mpi bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res 44:W410–W415
Baumer S, Ide T, Jacobi C, Johann A, Gottschalk G, Deppenmeier U (2000) The f420h2 dehydrogenase from methanosarcina mazei is a redox-driven proton pump closely related to nadh dehydrogenases. J Biol Chem 275:17968–17973
Boetius A, Wenzhofer F (2013) Seafloor oxygen consumption fuelled by methane from cold seeps. Nat Geosci 6:725–734
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, Amann R, Jorgensen BB, Witte U, Pfannkuche O (2000) A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623–626
Bowles M, Joye S (2011) High rates of denitrification and nitrate removal in cold seep sediments. ISME J 5:565–567
Bruggemann H, Falinski F, Deppenmeier U (2000) Structure of the f420h2: Quinone oxidoreductase of archaeoglobus fulgidus identification and overproduction of the f420h2-oxidizing subunit. Eur J Biochem 267:5810–5814
Capellagutiérrez S, Sillamartínez JM, Gabaldón T (2009) Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Cui M, Ma A, Qi H, Zhuang X, Zhuang G (2015) Anaerobic oxidation of methane: an "active" microbial process. MicrobiologyOpen 4:1–11
Cui HP, Su X, Chen F, Wei SP, Chen SH, Wang JL (2016) Vertical distribution of archaeal communities in cold seep sediments from the jiulong methane reef area in the south china sea. Biosci J 32:1059–1068
Delmont TO, Eren AM (2018) Linking pangenomes and metagenomes: the prochlorococcus metapangenome. PeerJ 6:e4320
Du MZ, Liu S, Zeng Z, Alemayehu LA, Wei W, Guo FB (2018a) Amino acid compositions contribute to the proteins' evolution under the influence of their abundances and genomic gc content. Sci Rep 8:7382
Du ZF, Zhang X, Luan ZD, Wang MX, Xi SC, Li LF, Wang B, Cao L, Lian C, Li CL, Yan J (2018b) In situ raman quantitative detection of the cold seep vents and fluids in the chemosynthetic communities in the south China Sea. Geochem Geophy Geosys 19:2049–2061
Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO (2015) Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3:e1319
Ettwig KF, Butler MK, Paslier DL, Pelletier E, Mangenot S, Kuypers MMM, Schreiber F, Dutilh BE, Zedelius J, Beer D, Gloerich J, Wessels HJCT, Alen T, Luesken F, Wu ML, Pas-Schoonen KT, Camp HJM, Janssen-Megens EM, Francoijs KJ, Stunnenberg H et al (2010) Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464:543–548
Feng D, Cheng M, Kiel S, Qiu JW, Yang QH, Zhou HY, Peng YB, Chen DF (2015) Using Bathymodiolus tissue stable carbon, nitrogen and sulfur isotopes to infer biogeochemical process at a cold seep in the South China Sea. Deep Sea Res PT I 104:52–59
Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT (2001) Acetyl-coa synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem 276:11420–11426
Gardner N, Champagne CP (2005) Production of Propionibacterium shermanii biomass and vitamin B12 on spent media. J Appl Microbiol 99:1236–1245
Glass JB, Yu H, Steele JA, Dawson KS, Sun S, Chourey K, Pan C, Hettich RL, Orphan VJ (2014) Geochemical, metagenomic and metaproteomic insights into trace metal utilization by methane-oxidizing microbial consortia in sulphidic marine sediments. Environ Microbiol 16:1592–1611
Graupner M, Xu H, White RH (2000) Identification of the gene encoding sulfopyruvate decarboxylase, an enzyme involved in biosynthesis of coenzyme m. J Bacteriol 182:4862–4867
Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, Yuan Z, Tyson GW (2013) Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500:567–570
Howell DM, Xu H, White RH (1999) (r)-citramalate synthase in methanogenic archaea. J Bacteriol 181:331–333
Huang Y, Gilna P, Li W (2009) Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25:1338–1340
Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC (2012) Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28:2223–2230
Kanehisa M, Goto S (2000) Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Knittel K, Losekann T, Boetius A, Kort R, Amann R (2005) Diversity and distribution of methanotrophic archaea at cold seeps. Appl Environ Micro 71:467–479
Krogh A, Brown M, Mian IS, Sjolander K, Haussler D (1994) Hidden markov models in computational biology. Applications to protein modeling. J Mol Biol 235:1501–1531
Kumar S, Babu BV (2006) A brief review on propionic acid: a renewal energy source. In: Proceedings of National Conference on Environmental Conservation (NCEC-2006). Birla Institute of Technology and Science, Indian, pp 459–464
Langmead B, Salzberg S (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359
Larsen HC, Mohn G, Nirrengarten M, Sun Z, Stock J, Jian Z, Klaus A, Alvarez-Zarikian CA, Boaga J, Bowden SA, Briais A, Chen Y, Cukur D, Dadd K, Ding W, Dorais M, Ferré EC, Ferreira F, Furusawa A, Gewecke A et al (2018) Rapid transition from continental breakup to igneous oceanic crust in the South China Sea. Nat Geosci 11:782–789
Levin LA (2005) Ecology of cold seep sediments: Interactions of fauna with flow, chemistry and microbes. Oceanogr Mar Biol 43:1–46
Liu F, Zhou HY, Wang CS, Li XY, Chen CB (2014) Chinese JIAOLONG’s first scientific cruise in 2013. J Ship Mechs 18(3):1–8
Liu L, Zhu Y, Li J, Wang M, Lee P, Du G, Chen J (2012) Microbial production of propionic acid from propionibacteria: current state, challenges and perspectives. Crit Rev Biotechnol 32:374–381
McGlynn SE (2017) Energy metabolism during anaerobic methane oxidation in anme archaea. Microbes Environ 32:5–13
Mehta AP, Abdelwahed SH, Mahanta N, Fedoseyenko D, Philmus B, Cooper LE, Liu Y, Jhulki I, Ealick SE, Begley TP (2015) Radical s-adenosylmethionine (sam) enzymes in cofactor biosynthesis: a treasure trove of complex organic radical rearrangement reactions. J Biol Chem 290:3980–3986
Miller LD, Russell MH, Alexandre G (2009) Diversity in bacterial chemotactic responses and niche adaptation. Adv Appl Microbiol 66:53–75
Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA (2013) Assembling single-cell genomes and mini-metagenomes from chimeric mda products. J Comput Biol 20:714–737
Oni OE, Friedrich MW (2017) Metal oxide reduction linked to anaerobic methane oxidation. Trends Microbiol 25:88–90
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) Checkm: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055
Quevillon ESV, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R (2005) Interproscan: protein domains identifier. Nucleic Acids Res 33:W116–120
Ragsdale SW, Pierce E (2008) Acetogenesis and the wood-ljungdahl pathway of CO2 fixation. Biochem Biophys Acta 1784:1873–1898
Scheller S, Yu H, Chadwick GL, McGlynn SE, Orphan VJ (2016) Artificial electron acceptors decouple archaeal methane oxidation from sulfate reduction. Science 351:703–707
Schulz TKF, Kluytmans JH (1983) Pathway of propionate synthesis in the sea mussel mytilus-edulis-l. Comp Biochem Phys B: Comp Biochem 75:365–372
Seemann T (2014) Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Shen L, Zhu Q, Liu S, Du P, Zeng J, Cheng D, Xu X, Zheng P, Hu B (2014) Molecular evidence for nitrite-dependent anaerobic methane-oxidising bacteria in the jiaojiang estuary of the east sea (China). Appl Microbiol Biotechnol 98:5029–5038
Silvestro D, Michalak I (2012) Raxmlgui: A graphical front-end for raxml. Organisms Div Evol 12:335–337
Skennerton CT, Chourey K, Iyer R, Hettich RL, Tyson GW, Orphan VJ (2017) Methane-fueled syntrophy through extracellular electron transfer: Uncovering the genomic traits conserved within diverse bacterial partners of anaerobic methanotrophic archaea. mBio 8:e00530-17
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The cog database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Tyree RW, Clausen EC, Gaddy JL (1991) The production of propionic-acid from sugars by fermentation through lactic-acid as an intermediate. J Chem Technol Bio 50:157–166
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF (2004) Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428:37–43
Wang F-P, Zhang Y, Chen Y, He Y, Qi J, Hinrichs K-U, Zhang X-X, Xiao X, Boon N (2013) Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J 8:1069–1078
Wang FP, Zhang Y, Chen Y, He Y, Qi J, Hinrichs KU, Zhang XX, Xiao X, Boon N (2014) Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J 8:1069–1078
Wang Y, Huang JM, Cui GJ, Nunoura T, Takaki Y, Li WL, Li J, Gao ZM, Takai K, Zhang AQ, Stepanauskas R (2019) Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ Microbiol 21:716–729
Wu Y, Qiu JW, Qian PY, Wang Y (2018) The vertical distribution of prokaryotes in the surface sediment of Jiaolong cold seep at the northern South China Sea. Extremophiles 22:499–510
Zdobnov EMAR (2001) Interproscan-an integrantion platform for the signature-recognition in interpro. Bioinformatics 17:847–848
Zhang Y, Su X, Chen F, Wang YY, Jiao L, Dong HL, Huang YY, Jiang HC (2012) Microbial diversity in cold seep sediments from the northern South China Sea. Geosci Front 3:301–316
Acknowledgements
We are grateful to Dr. JW. Qiu for the sample collection during a Jiaolong manned submersible dive. This study was supported by the National Key Research and Development Program of China (2018YFC0310005 and 2016YFC0302500), and the National Science Foundation of China (No. 31460001 and No. 41476104).
Author information
Authors and Affiliations
Contributions
WL analyzed the data and wrote the manuscript. YW completed the experiments. GZ and HH edited the manuscript. YW designed the experiments, analyzed the data and edited the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that there are no conflicts of interest.
Animal and human rights statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Edited by Chengchao Chen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, WL., Wu, YZ., Zhou, Gw. et al. Metabolic diversification of anaerobic methanotrophic archaea in a deep-sea cold seep. Mar Life Sci Technol 2, 431–441 (2020). https://doi.org/10.1007/s42995-020-00057-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-020-00057-9