
Abstract
Maize streak virus (MSV) continues to be a major biotic constraint for maize production throughout Africa. Concerning the quantitative nature of inheritance of resistance to MSV disease (MSVD), we sought to identify new loci for MSV resistance in maize using F2:3 population. The mapping population was artificially inoculated with viruliferous leafhoppers under screenhouse and evaluated for MSVD resistance. Using 948 DArT markers, we identified 18 quantitative trait loci (QTLs) associated with different components of MSVD resistance accounting for 3.1–21.4% of the phenotypic variance, suggesting that a total of eleven genomic regions covering chromosomes 1, 2, 3, 4, 5 and 7 are probably required for MSVD resistance. Two new genomic regions on chromosome 4 revealed the occurrence of co-localized QTLs for different parameters associated with MSVD resistance. Moreover, the consistent appearance of QTL on chromosome 7 for MSVD resistance is illustrating the need for fine-mapping of this locus. In conclusion, these QTLs could provide additional source for breeders to develop MSV resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amongst the maize virus diseases, Maize streak virus (MSV, genus Mastrevirus) still is a persistent concern that continues to cause significant yield loss in maize production areas across sub-Saharan Africa (SSA). MSV Disease (MSVD) was first reported in South Africa in the beginning of twentieth century and is now known to occur throughout Africa south to the Sahara and adjacent to the Indian Ocean islands (Fuller 1901; Harkins et al. 2009). MSV is transmitted by several species of leafhoppers belonging to the genus Cicadulina, especially C. mbila (Naudé) and C. triangular (Ruppel) in a persistent and circulative mode of transmission. Although MSVD incidence and severity depend on the virus inoculum sources, vector population and dynamics, stage of infection, surrounding agro-ecology and plant resistance to the disease (Bosque-Pérez 2000). Nonetheless, its incidence can lead up to 100% yield losses in susceptible maize cultivars if MSV outbreak occurs in the presence of drought or irregular early rains (Danson et al. 2006). The unpredictable vector survival and migratory patterns makes breeding for MSVD resistance, as well as its management under field conditions, extremely difficult (Danson et al. 2006).
Initial report describing the genetics of MSV resistance concluded the resistance to MSV to be quantitatively inherited (Gorter 1959). Contrarily, Storey and Howland (1967) suggested monogenic resistance with incomplete dominance. However, following the establishment of International Institute of Tropical Agriculture (IITA) at Ibadan, Nigeria, IITA scientists in collaboration with the International Maize and Wheat Improvement Center (CIMMYT) and national programs in SSA, initiated rigorous screening for MSV resistance under controlled artificial infestation facilities developed at IITA. This led to the development of the MSV resistant line IB32 which was derived from TZ-Y (Tropical Zea-Yellow) material (Efron et al. 1989). Subsequent, genetic study involving IB32 suggested quantitative inheritance of resistance to MSV conditioned by 2–3 genes with additive type of gene action (Kim et al. 1989). Similarly, Rodier et al. (1995) also suggested that multiple loci (major and minor) are associated with different levels of resistance to MSV (partial to complete) in maize.
With the advent of molecular markers, it has been well established that genetic resistance to MSV is governed by several loci depending upon the source material (Redinbaugh and Zambrano 2014). However, a major locus on chromosome 1, referred to as Msv1 accounted for 40–76% phenotypic variation, has been consistently reported in mapping populations derived from different MSV resistant lines (Welz et al. 1998; Kyetere et al. 1999; Pernet et al. 1999a, 1999b). This locus had shown additive or partial dominant gene action depending on the resistance source (Welz et al. 1998; Kyetere et al. 1999; Pernet et al. 1999a, b; Nair et al. 2015). Recently, the fine mapping of Msv1 locus enabled to delimit the region to 0.87 cM, harbouring candidate gene, GRMZM2G046848 which is a U-box domain containing tyrosine kinase family protein (Nair et al. 2015). Moreover, some additional quantitative trait loci (QTLs), with moderate to minor genetic effect for MSV resistance, have been also mapped on chromosomes 2, 3, 4, 6, 7, 9 and 10 but appear to be mostly either germplasm and/or environment specific (Redinbaugh and Zambrano 2014).
The deployment of maize cultivars resistant to MSV is the most preferred approach in SSA where approximately 55 million smallholder farmers depend on its cultivation. Thus, identification of new loci associated with MSV resistance in maize is highly relevant, because the over dependence on a single locus (Msv1) can pose a major threat to continue to develop maize cultivars with durable resistance to the virus in the SSA. In order to diversify the sources of genes for MSV resistance, we aimed to map loci conferring resistance to MSV in maize using a F2:3 population.
Materials and methods
Development of mapping population, inoculation for MSV infection and phenotyping
Among the maize inbreds previously screened for MSV at IITA-Ibadan, Nigeria, an inbred ((KU1414x9450)x9450)-15-2-1-BBB-1-B*11 (hereafter referred to as KU-R) was identified as having moderate resistance to MSV infection which displayed a reduction in symptom severity in the newly emerging leaves, resulting in high percent recovery from MSVD (Table 1). While the genotype (GT-MAS:GkxKU1414SRxGT-MAS:Gk)-8-1-2-4-B*12 (hereafter referred to as GT-S) showed relatively moderate level of susceptibility to MSV with very low recovery from infection under field conditions. Based on field analysis, both inbreds (KU-R as female and GT-S as male) were crossed to produce F1 hybrid in breeding nursery at IITA-Ibadan, Nigeria (7° 29′ 11.99″ N, 3° 54′ 2.88″ E, altitude 190 m). In order to develop a F2:3 mapping population, unselected F2 plants were self-pollinated (Veldboom et al. 1994). During the major crop season of 2017, a total of 105 F2:3 lines were phenotyped along with their parental lines for resistance to MSV under controlled artificial infestation in an insect-proof screenhouse at IITA-Ibadan, Nigeria. Each line was planted in pot and the pots were arranged in an alpha-lattice design with two replications each with eight plants per line per replication. A highly MSV susceptible line (Pool 16) was also grown alongside the test lines to verify that plants were properly inoculated by the viruliferous leafhopper vector. One-week old seedlings were artificially inoculated with a laboratory reared viruliferous leafhopper (C. triangular) colony in a screenhouse at 25–30 °C. Subsequently, visual evaluation of MSV symptoms on individual test plants was rated in F2:3 lines for MSVD severity using a scale of 1–5 (Fig. S1) following 2 weeks after inoculation (WAI) at weekly intervals for 6 weeks as described previously (Kim et al. 1989; Soto et al. 1982). Percent MSVD recovery was estimated as percent decrease in symptom severity at 6WAI compared to the maximum MSVD severity observed in the line during the 2 to 6WAI period. The symptom severity scores were also used to estimate the Area Under the Disease Progress Curve (AUDPC) (Shaner and Finney 1977).
DNA sampling and DArT genotyping
Samples were collected by punching leaves of each plant individually, and DNA extraction was performed from lyophilised samples using DArT protocol (www.diversityarrays.com). The DNA quality of each sample was monitored on 1% agarose gel and subsequently quantified using ND-1000 Spectrophotometer (Nanodrop Technologies). Library construction, sequencing and SNP calling was performed at the DArT Facility, Canberra, Australia. SNPs were called using DArTsoft analytical pipeline (http://www.diversityarrays.com/software.html#dartsoft). The sequenced reads were aligned with the sequence tag based in the maize meta-genome representation. A total of 3477 unimputed SNPs were successfully called within this F2:3 population.
Linkage map construction and QTL analysis
Each marker locus was tested for the expected allelic frequency of 0.5 by Χ2 tests in JoinMap4.0® (Van Ooijen 2006). The Kosambi mapping function was used to convert recombination values to map distances. The genetic map quality was assessed by comparing the positions of the mapped markers in each linkage group with their positions in the B73 reference genome version 4.0. Finally, the linkage map was constructed with 948 SNP markers (Table S1).
QTL identification was done using Inclusive composite interval Mapping using IciMapping v.4.1 software. IciMapping software was chosen due to its improved algorithm of CIM with an increased power to detect the QTLs as well as its capacity to reduce false detection rates and a lower biased QTL effect estimates (Manavalan et al. 2015). A QTL was considered as major QTL if it accounts for more than 10% phenotypic variance explained (PVE).
Statistical analysis
Analyses of variances were performed for all traits using PROC GLM in SAS software version 9.1 (SAS Institute 2003). Genotypes were considered as fixed effect while replications and blocks within replications as random effects. Data in percent were arcsine transformed before performing the analysis, but the original value was reported after back-transformation.
Results
F2:3 lines response to MSV inoculation
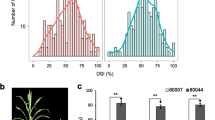
The susceptible check (Pool 16) and the parents exhibited 100% MSV incidence starting from 2WAI. At 3WAI, MSVD severity in the mapping population was at peak with the mean value of 4.8 at five-point scale (Fig. S2). Subsequently, the mapping population showed evidence of recovery. The inbred KU-R used as resistant parent displayed mean MSVD severity of 2.67 while the susceptible parent (GT-S) exhibited mean MSVD severity of 4.08 (Table 1). The differences in reaction to MSVD severity was much more pronounced between both parents at 4WAI (Table 1; Fig. S2). Under heavily diseased conditions, the resistant parent, KU-R, showed significant reduction in disease severity from 2WAI to 6WAI stage which is consistent with our previous observations under field conditions. In general, frequency distribution of MSVD severity showed significant skewness toward the susceptible parental mean (Table 1; Fig. S2). The broad sense heritability estimates for mean MSVD severity as well as specific stage (4, 5 and 6 WAI), percent MSVD recovery and AUDPC varied between 0.927 and 0.960.
Genetic map
A total of 23,198 SNP markers were generated by the maize DArTseq (1.0) array and filtering for high quality polymorphic SNPs between both parents reduced the number of SNPs to 3477. Of these SNP markers, 1763 SNP markers displaying minimal segregation distortion were considered for the map construction. Subsequently, after removing the completely linked markers with no more than 10% of missing data, 948 SNP markers remained, which could be assembled into stable framework order into 10 linkage groups corresponding to the number of haploid chromosomes (Fig. S3). The map spanned a cumulative distance of 5512 cM with an average inter-marker spacing of 5.90 cM and a maximum spacing of 33.8 cM (Table 2). Chromosome 1 had the highest number of markers (150), while chromosome 3 contained lowest number of markers (58). The length of linkage groups varied from 390.8 cM (Chr. 3) with a mean distance of 6.74 cM between adjacent markers to 827.1 cM (Chr.1) with a mean distance of 5.51 cM between adjacent markers. Chromosome 5 had the highest marker density (1 marker per 5.19 cM) while chromosome 9 contained the lowest marker density (1 marker per 6.81 cM). The positions of DArT markers on each chromosome were in general consistent with physical positions on B73 genome (Table S1).
QTL analysis for components of MSVD resistance
QTL analyses were performed on MSVD severity at different stages (4, 5 and 6 WAI) as well as mean MSVD severity, percent MSVD recovery and AUDPC using the genotypic data from 948 DArT markers. By using a LOD score of 2.5, Inclusive composite interval mapping under the additive model (ICIM-ADD) identified eighteen QTLs for MSVD resistance, ranging from 1 to 6 QTLs per parameter, six located on chromosome 4, four on chromosome 5 and two each on chromosome 1, 2, 3 and 7 (Table 3). QTL analysis revealed that a total of eleven genomic regions were involved in resistance to MSVD (Fig. S3).
For MSVD severity at 4WAI, two major QTLs (qMS4wai_4a and qMS4wai_4b) on chromosome 4 and one minor QTL (qMS4wai_5) on chromosome 5 were detected, accounting for 7.8–21.4% phenotypic variation with LOD score of 3.2 to 8.2. QTL, qMS4wai_4b had the largest effect on this trait, explaining 21.4% of the total phenotypic variation. Both parents contributed favourable alleles for this trait; the resistant parent (KU-R) allele at qMS4wai_4b and the susceptible parent (GT-S) alleles at qMS4wai_4a and qMS4wai_5 loci were associated with reduction in MSVD severity at 4WAI. Type of gene action observed was additive on qMS4wai_4a and qMS4wai_5, and overdominance on qMS4wai_4b. For MSVD severity at 5WAI, only a single major QTL on chromosome 2 was detected, accounting for 13.8% phenotypic variation with LOD score of 2.5. For this QTL, the allele from GT-S parent was favourable and type of gene action observed was partial dominance. While, three QTLS (qMS6wai_2, qMS6wai_3 and qMS6wai_7) were detected for MSVD severity at 6WAI. The QTL with the largest effect was qMS6wai_3 (PVE = 11.3%). For all the QTLs, the favourable alleles were contributed by GT-S and the effects of the alleles at the QTL involved in this trait were partial or over dominance.
For mean MSVD severity, five QTLs accounting for 7.3–18.2% PVE were detected on chromosomes 1, 3, 4 and 5. Of these QTLs, one QTL (qMMS_3) on chromosome 3 and two QTLs (qMMS_4a and qMMS_4b) on chromosome 4 were found to be major QTLs that could account for > 10% of the phenotypic variance. The resistant parent (KU-R) contributed the favourable allele at all the QTLs for this trait including the major QTLs qMMS_3 and qMMS_4b on chromosome 3 and 4 with additive and overdominance gene action, respectively, except qMMS_4a where GT-S contributed the allele to lower MSVD severity.
Interestingly, only single but major QTL (qPMR_7) located on chromosome 7 was identified for percent MSVD recovery, which could explain 12.2% phenotypic variance. The resistant parent (KU-R) contributed the favourable allele for the QTL with overdominance gene action.
Considering the QTLs associated with AUDPC, a total of five QTLs were found on chromosomes 1, 4 and 5 explaining between 7.0 and 17.8% of the phenotypic variance. Of these QTLs, two QTLs (qAUDPC_4a and qAUDPC_4b) detected on chromosome 4 were found to be major QTLs. The parent KU-R alleles at qAUDPC_1, qAUDPC_4b and qAUDPC_5a, and alleles from GT-S at qAUDPC_4a and qAUDPC_5b contributed to lower AUDPC. All type of gene action, additive to overdominance appeared to act for QTLs associated with AUDPC.
Epistatic interactions for components of MSVD resistance
The epistatic interactions for components of MSVD resistance were also analysed. As a result, 47 pairwise QTLs with epistatic interactions were identified for MSVD severity at 5 and 6 WAI, mean MSVD severity and percent MSVD recovery (Fig. 1). These epistatic QTLs (E-QTLs) were mapped to all the chromosomes (Table S2). Of these E-QTLs, most of E-QTLs were involved in MSVD severity at 5WAI (31 E-QTLs) and percent MSVD recovery (12 E-QTLs) accounting for 2.1–2.6% and 2.5–6.1% phenotypic variation, respectively. Interestingly, only two E-QTLs were detected for each MSVD severity at 6WAI and mean MSVD severity which explained 14.1–17.6% and 12.2–17.8% phenotypic variation, respectively. It is noteworthy that of the eleven genomic regions involved in direct effect QTL, five genomic regions were found to be involved in E-QTLs suggesting complexity of epistatic interactions for MSVD resistance. The major QTL associated with MSVD severity at 5WAI on chromosome 2, was also detected to be involved in interaction on chromosome 4 (0 cM) and 6 (205 cM) for mean MSVD severity which explained very high phenotypic variance (PVE; 12.2% and 17.8%, respectively).

Cyclic illustrations of epistatic interactions of QTLs associated with different components of MSVD resistance (a, b MSVD severity at 5WAI and 6WAI, respectively c mean MSVD severity and d percent MSVD recovery) in maize F2:3 population
Discussion
Genetic basis of quantitative resistance to MSVD was studied in a F2:3 population derived from a cross between two maize inbreds with contrasting resistance reactions to MSV. The frequency distribution of different parameters for MSVD resistance indicated that multiple genetic factors play an important role in resistance to MSV within the F2:3 lines. As shown in Table 1, none of the F2:3 lines were completely resistant suggesting the resistance conferred by parent KU-R provides partial protection against MSVD similar to that conferred by other MSVD resistant source such as CML202 (Asea et al. 2009). We identified twelve QTLs on chromosomes 1, 2, 3, 4, 5 and 7 for MSVD severity at different stages as well as mean MSVD severity, one QTL on chromosome 7 for percent MSVD recovery and five QTLs for AUDPC on chromosome 1, 4 and 5.
To date, the major QTL for MSV resistance on the chromosome 1 (Msv1) reported elsewhere using different maize genotypes was not detected in the present study (Welz et al. 1998; Kyetere et al. 1999; Pernet et al. 1999a, 1999b; Asea et al. 2009; Nair et al. 2015), indicating that Msv1 locus is not involved in the MSVD resistance in this population and the reaction of the resistant genotype seems to be associated with reduced virus replication. Nonetheless, some QTLs detected in this study corresponded well to the previously reported QTL for virus causing diseases including MSVD in maize (Table S3).
Most notable aspect of this study is that two genomic regions on chromosome 4 contained overlapping major QTLs for different components of MSVD resistance such as mean MSVD severity, MSVD severity at 4WAI and AUDPC. Nonetheless, these loci are most likely to be unique, as the chromosome 4 QTLs does not include any known gene or QTL related to MSVD in maize. Notably, a genomic region on chromosome 7 (12.8–33.0 Mbp) co-localized with a major QTL for percent MSVD recovery and a minor QTL for MSVD severity at 6WAI could be an effective target for breeding purposes. It is noteworthy to mention that this genomic region had also been previously reported to contain genetic factor for MSVD resistance (Pernet et al. 1999a). The consistently detected QTL for MSVD suggested the qPMR_7 locus needs to be fine mapped as it encompasses a quite large region (~ 20 Mb). Moreover, the minor QTLs detected on chromosome 1 (275.7–279.1 Mbp), 2 (15.6–17.6 Mbp) and 5 (199.5–210.8 Mbp) for MSVD resistance also showed good consistency with previous results of Pernet et al. (1999a, b).
Epistatic interactions are quite common and have been previously reported to be an important component of the genetic basis of disease resistance in maize including viral diseases (Xia et al. 1999; Wu et al. 2007; Jones et al. 2011). Of the epistatic loci detected in the present study, two epistatic QTLs each for MSVD severity at 6WAI and mean MSVD severity explaining high phenotypic variation suggest important role of epistatic interactions in MSVD resistance. Nonetheless, the power to detect epistasis varies with the size of the population, therefore, it is possible that effects of epistatic QTLs observed here might be overestimated when compared to using a larger population size (Carlborg and Haley 2004). Recently, Zambrano et al. (2014) also reported major interaction explaining up to 28% more of the total variance than the sum of their individual QTL effects for SCMV resistance in maize.
In general, it has been seen that loci conferring resistance to pests and diseases, including viruses, are often clustered in the maize genome which may be due to the numerous factors such as gene duplication, unequal crossing-over, insertion of transposons, effect of selection and recombination over time (Friedman and Baker 2007; Gururani et al. 2012; Lozano et al. 2012). In agreement with previous studies, six of the eleven genomic regions identified in this study also have been previously reported for either MSV or for other maize diseases including virus causing diseases such as SCMV (Xia et al. 1999; Zhang et al. 2003; Gustafson et al. 2018), MStV (Dintinger et al. 2005), MRDD (Luan et al. 2012) and MLND (Gowda et al. 2018) which clearly indicate the presence of gene clusters influencing resistance to multiple diseases in maize. Nevertheless, there is no evidence that they could be considered as the same QTL, because the underlying mechanisms are still unknown. The feature of broad-spectrum resistance makes the stacking of such major QTL with Msv1 locus appealing in practical applications because the divergent strains have biological significance in resistance to MSV and these QTL could have large effects in other germplasms.
Conclusions
Breeding resistant maize cultivars is the most cost-effective way to minimize yield losses from MSVD in sub-Saharan Africa. Thus, the main idea of mapping MSVD associated loci is to identify alternate source of Msv1 locus. This may greatly facilitate the stacking of the novel MSV QTLs to the lines relying on only Msv1 locus for MSVD resistance through marker-assisted selection. The two genomic regions on chromosome 4 having co-localized major QTLs seems to be novel, but further experiments are needed to validate their consistency in different genetic backgrounds. Other major QTLs, including the one on chromosome 7, coincided with the QTLs identified in previous studies for virus caused diseases including MSV in maize, suggesting the presence of gene clusters in these regions for resistance to virus diseases. The fine-mapping of major QTL consistently detected on chromosome 7 may lead to the discovery of novel MSV resistance mechanisms in maize. Finally, our result suggests that the QTL region identified on chromosome 4 and 7 may be exploited in breeding programs to develop maize varieties with durable resistance to MSV in Africa.
References
Asea G, Vivek BS, Bigirwa G, Lipps PE, Pratt RC (2009) Validation of consensus quantitative trait loci associated with resistance to multiple foliar pathogens of maize. Phytopathology 99(5):540–547. https://doi.org/10.1094/PHYTO-99-5-0540
Bosque-Pérez NA (2000) Eight decades of maize streak virus research. Virus Res 71(1–2):107–121. https://doi.org/10.1016/S0168-1702(00)00192-1
Carlborg O, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5(8):618. https://doi.org/10.1038/nrg1407
Danson J, Lagat M, Ininda J, Kimani M (2006) Application of simple sequence repeats (SSRs) markers to study the resistance of locally adapted maize hybrids to damaging maize streak virus disease. Afr J Biotechnol 5(16):1430–1434
Dintinger J, Verger D, Caiveau S, Risterucci AM, Gilles J, Chiroleu F, Courtois B, Reynaud B, Hamon P (2005) Genetic mapping of maize stripe disease resistance from the Mascarene source. Theor Appl Genet 111(2):347–359. https://doi.org/10.1007/s00122-005-2027-3
Efron Y, Kim SK, Fajemisin JM, Mareck JH, Tang CY, Dabrowski ZT, Rossel HW, Thottappilly G, Buddenhagen IW (1989) Breeding for resistance to maize streak virus: a multidisciplinary team approach. Plant Breed 103(1):1–36. https://doi.org/10.1111/j.1439-0523.1989.tb00347.x
Friedman AR, Baker BJ (2007) The evolution of resistance genes in multi-protein plant resistance systems. Curr Opin Genet Dev 17(6):493–499. https://doi.org/10.1016/j.gde.2007.08.014
Fuller C (1901) Mealie variegation. 1st Report of the Government Entomologist, Natal, 1899–1900
Gorter GJMA (1959) Breeding maize for resistance to streak. Euphytica 8(3):234–240. https://doi.org/10.1007/BF00039365
Gowda M, Beyene Y, Makumbi D, Semagn K, Olsen MS, Bright JM, Das B, Mugo S, Suresh LM, Prasanna BM (2018) Discovery and validation of genomic regions associated with resistance to maize lethal necrosis in four biparental populations. Mol Breed 38(5):66. https://doi.org/10.1007/s11032-018-0829-7
Gururani MA, Venkatesh J, Upadhyaya CP, Nookaraju A, Pandey SK, Park SW (2012) Plant disease resistance genes: current status and future directions. Physiol Mol Plant Pathol 78:51–65. https://doi.org/10.1016/j.pmpp.2012.01.002
Gustafson TJ, de Leon N, Kaeppler SM, Tracy WF (2018) Genetic analysis of sugarcane mosaic virus resistance in the wisconsin diversity panel of maize. Crop Sci 58(5):1853–1865. https://doi.org/10.2135/cropsci2017.11.0675
Harkins GW, Martin DP, Duffy S, Monjane AL, Shepherd DN, Windram OP, Owor BE, Donaldson L, Van Antwerpen T, Sayed RA, Flett B (2009) Dating the origins of the maize-adapted strain of maize streak virus, MSV-A. J Gen Virol 90(12):3066–3074. https://doi.org/10.1099/vir.0.015537-0
Jones MW, Boyd EC, Redinbaugh MG (2011) Responses of maize (Zea mays L.) near isogenic lines carrying Wsm1, Wsm2 and Wsm3 to three viruses in the Potyviridae. Theor Appl Genet 123:729–740. https://doi.org/10.1007/s00122-011-1622-8
Kim SK, Efron Y, Fajemisin JM, Buddenhagen IW (1989) Mode of gene action for resistance in maize to maize streak virus. Crop Sci 29(4):890–894. https://doi.org/10.2135/cropsci1989.0011183X002900040009x
Kyetere DT, Ming R, McMullen MD, Pratt RC, Brewbaker J, Musket T (1999) Genetic analysis of tolerance to Maize streak virus in maize. Genome 42:20–26. https://doi.org/10.1139/g98-099
Lozano R, Ponce O, Ramirez M, Mostajo N, Orjeda G (2012) Genome-wide identification and mapping of NBS-encoding resistance genes in Solanum tuberosum group phureja. PLoS ONE 7(4):e34775. https://doi.org/10.1371/journal.pone.0034775
Luan J, Wang F, Li Y, Zhang B, Zhang J (2012) Mapping quantitative trait loci conferring resistance to rice black-streaked virus in maize (Zea mays L.). Theor Appl Genet 125(4):781–791. https://doi.org/10.1007/s00122-012-1871-1
Manavalan LP, Prince SJ, Musket TA, Chaky J, Deshmukh R, Vuong TD, Song L, Cregan PB, Nelson JC, Shannon JG, Specht JE (2015) Identification of novel QTL governing root architectural traits in an interspecific soybean population. PLoS ONE 10(3):e0120490. https://doi.org/10.1371/journal.pone.0120490
Nair SK, Babu R, Magorokosho C, Mahuku G, Semagn K, Beyene Y, Das B, Makumbi D, Kumar L, Olsen M, Boddupalli PM (2015) Fine mapping of Msv1, a major QTL for resistance to Maize Streak Virus leads to development of production markers for breeding pipelines. Theor Appl Genet 128:1839–1854. https://doi.org/10.1007/s00122-015-2551-8
Pernet A, Hoisington D, Dintinger J, Jewell D, Jiang C, Khairallah M, Letourmy P, Marchand JL, Glaszmann JC, González de León D (1999a) Genetic mapping of maize streak virus resistance from the Mascarene source. II. Resistance in line CIRAD390 and stability across germplasm. Theor Appl Genet 99:540–553. https://doi.org/10.1007/s001220051267
Pernet A, Hoisington D, Franco J, Isnard M, Jewell D, Jiang C, Marchand JL, Reynaud B, Glaszmann JC, González de León D (1999b) Genetic mapping of maize streak virus resistance from the Mascarene source. I. Resistance in line D211 and stability against different virus clones. Theor Appl Genet 99:524–539. https://doi.org/10.1007/s001220051266
Redinbaugh MG, Zambrano JL (2014) Control of virus diseases in maize. In: Smith KM, Lauffer MA (eds) Advances in virus research, vol 90. Academic Press, Cambridge, pp 391–429. https://doi.org/10.1016/B978-0-12-801246-8.00008-1
Rodier A, Assie J, Marchand JL, Herve Y (1995) Breeding maize lines for complete and partial resistance to maize streak virus (MSV). Euphytica 81(1):57–70. https://doi.org/10.1007/BF00022459
SAS Institute (2003) The SAS system for Windows V. 9.1. SAS Institute, Cary, North Carolina
Shaner G, Finney RE (1977) The effect of nitrogen fertilization on the expression of slow-mildewing resistance in Knox wheat. Phytopathology 67(8):1051–1056. https://doi.org/10.1094/Phyto-67-1051
Soto PE, Buddenhagen IW, Asnani VL (1982) Development of streak virus-resistant maize populations through improved challenge and selection methods. Ann Appl Biol 100(3):539–546. https://doi.org/10.1111/j.1744-7348.1982.tb01420.x
Storey HH, Howland AK (1967) Inheritance of resistance in maize to the virus of streak disease in East Africa. Ann Appl Biol 59:429–436. https://doi.org/10.1111/j.1744-7348.1967.tb04459.x
Stuber CW, Edwards M, Wendel JF (1987) Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Sci 27(4):639–648. https://doi.org/10.2135/cropsci1987.0011183x002700040006x
Van Ooijen JW (2006) JoinMap 4. Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, p 33
Veldboom LR, Lee M, Woodman WL (1994) Molecular marker-facilitated studies in an elite maize population: I. Linkage analysis and determination of QTL for morphological traits. Theor Appl Genet 88(1):7–16. https://doi.org/10.1007/BF00222387
Welz HG, Schechert A, Pernet A, Pixley KV, Geiger HH (1998) A gene for resistance to the maize streak virus in the African CIMMYT maize inbred line CML202. Mol Breed 4:147–154. https://doi.org/10.1023/A:1009602620244
Wu J, Ding J, Du Y, Xu Y, Zhang X (2007) Genetic analysis and molecular mapping of two dominant complementary genes determining resistance to Sugarcane mosaic virus in maize. Euphytica 156:355–364. https://doi.org/10.1007/s10681-007-9384-8
Xia X, Melchinger AE, Kuntze L, Lubberstedt T (1999) Quantitative trait loci mapping of resistance to sugarcane mosaic virus in maize. Phytopathology 89(8):660–667. https://doi.org/10.1094/PHYTO.1999.89.8.660
Zambrano JL, Jones MW, Brenner E, Francis DM, Tomas A, Redinbaugh MG (2014) Genetic analysis of resistance to six virus diseases in a multiple virus-resistant maize inbred line. Theor Appl Genet 127(4):867–880. https://doi.org/10.1007/s00122-014-2263-5
Zhang SH, Li XH, Wang ZH, George ML, Jeffers D, Wang FG, Liu XD, Li MS, Yuan LX (2003) QTL mapping for resistance to SCMV in Chinese maize germplasm. Maydica 48:307–312
Acknowledgements
We acknowledge assistance of IITA field and laboratory technical staff. This work was supported by the CGIAR Research Program on Maize, and Stress Tolerant Maize for Africa project (Grant No. OPP1134248) funded by Bill and Melinda Gates Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Mohan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
42976_2020_20_MOESM2_ESM.jpg
Figure S2. Frequency distribution of mean MSVD severity (A) and at different stages after inoculation [6WAI (B), 5WAI (C), 4WAI (D), 3WAI (E) and 2WAI (F)], percent MSVD recovery (G) and AUDPC (H) in a population of 93 F2:3 lines derived from cross between KU-R and GT-S. Note: Inverted triangles indicate values/ratings of the parental lines. Inverted white and black triangles represents MSVD susceptible (GT-S) and resistant (KU-R) parents, respectively (JPEG 213 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Garcia-Oliveira, A.L., Menkir, A., Kumar, P.L. et al. Quantitative trait loci mapping for resistance to maize streak virus in F2:3 population of tropical maize. CEREAL RESEARCH COMMUNICATIONS 48, 195–202 (2020). https://doi.org/10.1007/s42976-020-00020-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42976-020-00020-5