
Abstract
Antimony has become an increasingly critical element in recent years, due to a surge in industrial demand and the Chinese domination of primary production. Antimony is produced from stibnite ore (Sb2O3) which is processed into antimony metal and antimony oxide (Sb2O3). The industrial importance of antimony is mainly derived from its use as flame retardant in plastics, coatings, and electronics, but also as decolourizing agent in glass, alloys in lead-acid batteries, and catalysts for the production of PET polymers. In 2014, the European Commission highlighted antimony in its critical raw materials report, as the element with the largest expected supply–demand gap over the period 2015–2020. This has sparked efforts to find secondary sources of antimony either through the recycling of end-of-life products or by recovering antimony from industrial process residues. Valuable residues are obtained by processing of gold, copper, and lead ores with high contents of antimony. Most of these residues are currently discarded or stockpiled, causing environmental concerns. There is a clear need to move to a more circular economy, where waste is considered as a resource and zero-waste valorization schemes become the norm, especially for rare elements such as antimony. This paper gives a critical overview of the existing attempts to recover antimony from secondary sources. The paper also discusses the possibility of waste valorization schemes to guarantee a more sustainable life cycle for antimony.
Similar content being viewed by others


Introduction
The history of antimony goes back to as early as 3100 BC, when it was used by the Egyptians as cosmetic in the form of black Sb2S3 [1]. Antimony was later used by alchemists in the form of antimony oxychloride SbOCl, which acted as a powerful emetic and thus became known as “mercury of life” or “algarot” [2]. The etymology of the current name “antimony” is still subject of discussion. Some believe it comes from the Greek word antimonos (“never found alone”), others have suggested it is derived from the greek word anthos (“flower”), due to the petal-like appearance of stibnite ore, and some even argued that it is derived from the Greek word anti-monachos or the French word antimoine (“against monks”), which could refer to the poisonous properties of antimony and the fact that many early alchemists were monks [1, 2]. Despite its long history, antimony metal was isolated and identified as late as the 16th century. Today, antimony is used in a wide variety of products and processes. It is mainly produced from stibnite ore (Sb2S3), but also occurs as oxide (Sb2O3), and as antimonides and sulphoantimonides of metals like lead, copper, zinc, silver, and gold [3]. Antimony is mainly used in the form of Sb2O3, as flame retardant in plastics, coatings, and electronics, due to its synergetic effect with halogenated flame retardants, which minimizes the amount of halogenated flame retardant required [4]. This application takes up the majority of the world’s antimony production [4, 5]. Antimony is also used in catalysts for the production of polyethylene terephthalate (PET) polymers and as additive in glass in the form of sodium antimonite, which acts as a decolourizing agent for optical glass in cameras, photocopiers, binoculars, and fluorescent light tubes [3, 6]. Antimony metal is used as a hardener in lead alloys such as the lead electrodes in lead-acid (LA) batteries [1, 7]. The average antimony content of automotive battery alloys has declined from 7 to 1.6 % in recent years, as Ca, Al, and Sn alloys have been used as replacements. However, the increasing car sales partly compensate for the declining use of antimony in LA batteries [5, 7]. Other minor uses include paint pigments (Sb2O3, Sb2S3, Sb2S5), semiconductors (e.g., AsSb, GaSb, InSb), IR-reflecting camouflage paints (Sb2S3), matchboxes (Sb2S3), and vulcanizing agents in the production of red rubber (Sb2S5) [2, 7]. Antimony compounds are also used in certain phosphors (for instance the halophosphate lamp phosphors), pesticides, ammunition, and medicine [7]. Overall, it is estimated that the global antimony consumption is distributed to flame retardants 52 %, lead alloys and lead-acid batteries 38 %, catalysts for the production of polyethylene terephthalate (PET) 6 %, and chemicals, ceramics, and glass less than 3 %, down from 13 % in 2000 [3]. More detailed overviews of the antimony market and applications can be found in industry reports and government reports [3, 5].
Antimony mining is currently dominated by China (≈78 % of global production), which also holds the largest reserves (Table 1) [1]. The dependence on China, combined with the strong industrial demand for antimony, has raised concerns over the supply security, especially since the abundancy of antimony in the earth’s crust is quite low (0.2 ppm) and current reserves (1,800,000 t) allow for only 10–11 more years of production at the current speed (Table 1) [8].
Figure 1 shows the distribution of primary and secondary production as well as the portion of illegal (non-reported) primary mining based on a 2011 consulting report [3]. Despite the presence of some deposits, no antimony is currently mined in Europe or in the U.S.A. [1, 7].

Distribution of antimony ore production in 2010 (196,484 t), using data compiled by Roskill [3]
These observations have led the European Commission to highlight antimony as a critical raw material in 2014, with an expected supply–demand gap exceeding 10 % over the period 2015–2020, which is the highest amongst all critical metals [9]. The processing of antimony ores and the production of antimony metal is also concentrated in China, due to its high smelting capacity. Most of the antimony industry in Europe and the U.S.A. is therefore dependent on the import of Chinese antimony metal and is focused on the production of high-purity products and applications.
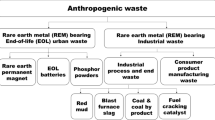
The aim of this paper is to give an overview of antimony-containing waste streams and to analyze their potential as secondary sources of antimony. Furthermore, the existing valorization methods are discussed as well as the environmental hazard associated with antimony. Finally, an overview is given of the existing water purification and air treatment methods to recover traces of antimony and avoid unwanted release of antimony into the environment. Secondary supply, through recycling and valorization of industrial residues could be a solution to ensure a more secure long-term supply of antimony [9]. Secondary production of antimony could also make Europe and the U.S.A. less reliable on Chinese antimony, thus lowering geopolitical risks. Roskill Consulting estimated that in 2010 around 20 % of the global antimony supply originated from secondary production [3]. Today, secondary production of antimony is mainly restricted to the recycling of antimony-containing lead alloys from lead-acid battery recycling plants [3]. However, interesting future secondary sources could include industrial residues (e.g., mine tailings, process residues, manufacturing scrap) from the production of lead, copper, gold, and antimony [7]. The large production volumes, especially for copper and lead, mean that these residues have the potential to replace a large portion of the primary antimony production [7]. Secondary antimony sources also include end-of-life products such as lead-acid batteries, plastics with antimony-containing flame retardants, antimony-containing glass, and phosphor powders from spent fluorescent lamps [7]. At this moment, only lead-acid batteries are being recycled on a large scale [10, 11]. Antimony could also be recycled from spent fluid catalytic cracking (FCC) catalysts or from catalysts used for the production of PET polymers [12]. Finally, efforts have been made to recover antimony from landfills and municipal solid waste incineration (MSWI) ashes [13–15]. A schematic overview of the antimony life cycle, with its waste streams and possible recycling routes, is depicted in Fig. 2.

Schematic overview of the antimony life cycle, which includes (1) the main value chain between primary mining and final disposal, (2) the major waste streams, and (3) the possible secondary antimony production routes through recycling and recovery of antimony-containing waste (Color figure online)
Recovery from Industrial Residues
Antimony Production
The primary production and metallurgy of antimony has been reviewed by Anderson (2012) [1]. However, in order to discuss the secondary production of antimony from industrial residues, a short overview must be given here of the main production routes, process residues, and metal-containing waste streams. The main pyro- and hydrometallurgical pathways to produce antimony metal and antimony oxide (Sb2O3), are shown in Fig. 3.

Primary production of antimony through pyrometallurgical and hydrometallurgical methods
Hydrometallurgical methods are based on two steps: leaching (alkaline sulfide or acidic chloride system), followed by the electrodeposition of antimony metal at the cathode, or hydrolysis with NaOH or NH4OH to produce Sb2O3 [1]. At this moment, mainly pyrometallurgical processes are used, but hydrometallurgical processes based on alkaline sulfide leaching have been employed industrially in the former Soviet Union, China, Australia, and the United States [1]. Alkaline sulfide technology has the advantage of having a high selectivity toward antimony, and having less issues with corrosion. However, a considerable amount of research and pilot-scale work has been undertaken to utilize chloride-based technology [1, 16, 17]. A more detailed overview of the pyrometallurgical pathways and process residues is given in Fig. 4.

Pyrometallurgical pathways (blue) and metal-containing waste streams (red) for the primary production of antimony from stibnite ore. The different process residues can be used as secondary antimony sources and fed back into the blast furnace (green) (Color figure online)
A stibnite concentrate is first produced by grinding, milling, flotation, and gravity concentration [1]. These steps are usually carried out close to the mining site, and result in Sb-containing mine tailings. Depending on the grade of antimony in the concentrate, different pyrometallurgical methods can be used to extract antimony [1]. For low-grade ores (5–25 % Sb), oxide volatilization is used. The ores are roasted at 1000 °C, and the volatile Sb2O3 is recovered. Intermediate grade ores (25–40 % Sb), and Sb-rich residues, slags, mattes, and flue dusts are smelted in a blast furnace at 1300–1400 °C which produces antimony metal, SO2 gas, and a slag. Sb-rich ores (45–60 % Sb) can be treated by liquation and iron precipitation. Liquation consists of heating the ore to 550–600 °C under a reducing atmosphere, to extract liquid Sb2S3 from the ore. This product is called crude, liquated, or needle antimony. The liquation residue (12–30 % Sb) can be treated to further improve the recovery of antimony. The iron precipitation method is used to produce antimony metal from concentrated antimony ores or from the crude antimony issued from the liquation process. In this process, scrap iron is added to molten Sb2S3 to displace the antimony in order to form antimony metal and a matte which contains iron sulfide. The reduction of Sb2O3 to antimony metal is done with charcoal in a reverberatory furnace (1200 °C), with the formation of a slag. The loss of antimony by volatilization is high (12–20 %), meaning that the flue dusts must also be caught and then reprocessed in a blast furnace. Finally, refining steps are needed to obtain pure antimony metal (e.g., arsenic removal with NaOH). This creates arsenate- and antimonate-containing residues. Highly pure Sb2O3 is usually obtained by oxidation of technical grade antimony metal (99.0–99.8 % pure), followed by a refuming process (volatilization + condenzation) [2]. These oxidation processes produce dusts and residues, which can also contain important quantities of antimony. It is clear from Fig. 4 that antimony production creates many residues. The composition of these residues is highly variable on the type of feed material and the process parameters, but some representative compositions are given in Table 2.
In some countries, these residues are stockpiled or landfilled, causing metal pollution in the areas around antimony mining sites and processing plants [18, 22–24]. A better approach is to try to immobilize these waste residues by applying various stabilization, solidification, and geopolymerization techniques [22]. However, a better solution is to valorize these residues, thus creating secondary sources of antimony. This increases the extraction efficiency of antimony from the initial ore and also eliminates or reduces the need to discard residues containing toxic metals [18]. Guo et al. [18] characterized the process residues (e.g., slags, flue dusts) from an antimony smelting plant in Xikuangshan area in China (the “antimony capital”). They found large amounts of antimony in the flue dust of the blast furnace (23.4 wt%) and in the residue from the arsenic removal process (37.8 wt%). The leaching of the different residues was studied in an effort to remove the antimony and other metals (e.g., Hg, Pb, As, Cr) from these waste residues. Fuxu et al. investigated the use of a three-phase fluidized bed reactor for arsenic removal from Sb-refining residue (20–40 % Sb, 3–5 % As) [21]. Their set-up achieved a 97 % removal rate of arsenic, leaving behind a valuable antimony concentrate. Kequiang et al. [21] prepared Sb2O3 (98.50 % pure) from Sb-containing slag, using a vacuum evaporation method. The slag was placed in the vacuum chamber and heated (893–1073 K) in order to selectively evaporate the volatile Sb2O3. Luo et al. [20] studied the recovery of gold and antimony from antimony smelting slag by direct reduction in an electric furnace. Using this method, the antimony content in the slag was reduced from 32 to <1 wt%. During the pyrometallurgical production of antimony oxide from antimony metal, waste residues are also created, which contain alloys of antimony and other base and precious metals. Anderson [7] studied the recovery of antimony from these pyrometallurgical production residues. Using alkaline sulfide leaching, 99.5 % of antimony was recovered.
At the same time, research is still ongoing to optimize the primary production of antimony from stibnite ore and to reduce the amount of (toxic) waste streams [7, 16, 25, 26]. Yang et al. [25], for example, developed a low-temperature sulfur-fixing smelting process, using a NaOH–Na2CO3 flux with ZnO to fix the sulfur as ZnS. This approach reduces SO2 emissions (98.61 % sulfur-fixing) and results in an antimony recovery rate of 97.07 % in the form of antimony metal. Ye et al. [26] investigated a similar system, using a eutectic Na2CO3–NaCl molten salt and ZnO as a sulfur-fixing agent. Under optimum conditions, the average recovery ratio of antimony reached 92.88 %. Also of interest, are the efforts to recover antimony from a low-grade or complex stibnite ore, as well as from secondary antimony minerals. Yang and Wu [16] developed a new hydrometallurgical process to recover antimony from complex stibnite concentrate. They dissolved antimony using a chlorination–oxidation procedure to obtain an SbCl3 solution. Using electrodeposition, hydrolysis, hydrolysis–smelting, or hydrolysis–washing, they obtained cathode antimony metal (99.98 %), Sb2O3 (99.9 %), crude antimony metal (99 %), or SbOCl (99.9 %), respectively. Gök (2014) published a hydrometallurgical method for the catalytic production of antimonate from stibnite concentrate [27]. Alkaline leaching was used to extract the antimony, followed by the oxidation of Sb(III) in a hydroquinone-catalyzed alkaline electrolyte, which precipitates antimony as sodium hydroxyantimonate NaSb(OH)6 (with 90 % recovery).
Besides stibnite, researchers are also investigating other, complex antimony minerals. An overview of complex antimony minerals and their mineralogy has been compiled by Roper et al. [28]. Complex antimony minerals often require adapted flowsheets in order to ensure the recovery of antimony and the other main metal constituents. The last primary producer of antimony in the U.S.A. (Sunshine Mining & Refining Company) was mining the complex copper-silver-antimony sulfide: freibergite (Cu,Ag)12Sb4S13 [1]. This mine produced both silver and antimony concentrates until its closure in 2001. Other interesting Sb-containing minerals are tetrahedrite (Cu,Fe)12Sb4S13 and jamesonite Pb4FeSb6S14. Baláž et al. [29] investigated the leachability of antimony and arsenic from tetrahedrite and jamesonite with alkaline sulfide leaching. Awe and Sandström [30] also studied the selective leaching of arsenic and antimony from a tetrahedrite-rich complex sulfide concentrate and concluded that by removing these elements, a valuable copper concentrate was obtained, which could be used as a feedstock for copper smelting. Yang et al. [31] developed a flowsheet for the recovery of antimony from a low-grade jamesonite concentrate (Pb4FeSb6S14). They used sodium sulfide to dissolve the antimony as sodium thioantimonate (Na3SbS4) and subsequently used air oxidation to produce sodium pyroantimonate. (Na2H2Sb2O7). Duchao et al. [32] improved this process by using pressured air oxidation. Under optimum conditions, the precipitation ratio of antimony was 99.38 %. Gold, copper, and lead production are also associated with large amounts of antimony impurities, which end up in the process residues.
Gold Production
Gold-bearing sulfide ores often contain important amounts of antimony (Table 3). Kyle et al. [38, 39] made a series of reports on the occurrence and deportment of trace elements (Sb, Bi, Se, Te, As, Hg, Cd, Pb) in gold processing. They estimated that in Australia alone, total emissions of antimony (air, water, land) reach as much as 3.8 t/year during mining and 8.7 t/year during metal manufacturing. This represents a significant loss of valuable antimony and it is a challenge to find better processing technologies which can minimize the loss of antimony by valorizing the different gold-mining residues and waste streams.
These “refractory” gold ores are resistant against the standard gold recovery method of cyanidation and carbon adsorption, because the sulfide mineral traps the gold particles, making it difficult for the cyanide leach solution to complex with the gold [2, 40]. Pretreatment is therefore required to make the cyanidation effective. Different pretreatment options exist: chemical leaching, roasting, bacterial oxidation, pressure oxidation, and ultrafine grinding [2, 40]. The choice of pretreatment process depends on the type of ore and the impurities present in the ore and may be preceded by a flotation concentration step. Depending on the concentrations of gold and antimony, different categories of Au-Sb-containing sulfide ores can be distinguished: (1) ores in which gold is the main valuable component and antimony is a harmful impurity, (2) gold-bearing ores in which the amount of antimony is considerable and for which it is economically viable to recover antimony as a commercial byproduct, (3) antimony ores in which gold is a minor accompanying element, the recovery of which could increase the economic efficiency of the process [38, 41]. For the ores of group (1) and (3), standard gold-processing and antimony-processing technology is applied, respectively. The recovery of traces of gold and antimony from these ores is only interesting for waste products, as this does not cause losses in the recovery of the main metal. The processing of the second group of ores (2), usually requires changes in the flowsheet in order to ensure the recovery of both the gold and antimony fraction. For this type of ores, alkaline sulfide leaching is a preferred pretreatment method as it isolates antimony and simultaneously increases the recovery rate of gold in the consequent cyanidation step (Fig. 5).

Schematic visualization of the pretreatment of refractory Au-Sb sulfide ore with alkaline sulfide leaching. The pretreatment method improves the cyanidation of gold and allows the recovery of antimony [40]
Ubaldini et al. [40] used alkaline sulfide leaching as a pretreatment step, to remove antimony from a gold-bearing stibnite ore. Antimony was then recovered by electrodeposition. Results showed that this pretreatment improved the recovery of gold in the consequent cyanidation process from 30 to 75 %. Celep et al. [35, 42] studied similar antimonial refractory gold and silver ores. Their findings confirmed that alkaline NaOH and Na2S leaching were appropriate pretreatment methods to recover antimony prior to the conventional cyanidation for the recovery of the noble metals. Using this approach, 85 % of antimony could be leached out and during the subsequent cyanidation step, silver recovery rates increased from less than 18 % up to 90 % and gold extraction was also enhanced by 20–30 %. Solozhenkin et al. (2010) investigated the complex antimony ores and gold-antimony concentrates, found in the Sarylakhsky and Sentachansky deposits in Russia [34, 43]. They developed different hydrometallurgical processes to treat these ores and produce both gold and antimony concentrates with antimony recovery rates of 95–98 %. The studied methods include antimonite flotation, sodium dimethyldithiocarbamate treatment, and bio-treatment of the ores. The authors also introduced a processing plant for Sb-Au-bearing alloys and demonstrated the successful electrolytic refining and production of cathode antimony and noble metal slurry [39, 43]. Kanarskii et al. [38] investigated the concentration and isolation of antimony and arsenic in Sb-As-bearing gold ores by flotation. They demonstrated the flotation separation of antimonite and arsenopyrite into separate products, thus increasing process efficiency and profit by improving the gold recovery rate and reducing the consumption of cyanide. Karimi et al. [36] investigated the influence of different pretreatment methods (H2O2/air oxidation, roasting, and HNO3/HCl leaching) on the cyanidation of Sb-rich gold ores and the movement of antimony and impurities. Saleh et al. [37], examined the recovery of Au, Sb, and Sn from gold electrorefining slime. The solid waste was dissolved in HCl (2 M), before extracting the metal ions with the quaternary ammonium chloride extractant Aliquat® 336.
Copper Production
Copper is mainly produced from copper ores such as chalcopyrite, CuFeS2, which often contain antimony, arsenic, and bismuth as impurities [2]. These impurities must be removed and therefore end up in various residues. The formation of the residues is discussed here, as well as the recovery of antimony from these residues. In the past, the copper concentrate (20–40 % Cu) was first roasted and then introduced in blast furnaces or reverberatory furnaces, but nowadays direct smelting is favored using for example Outokumpu Flash furnaces or Isasmelt furnaces [2, 44]. During smelting, silica is added as a flux to remove iron as an iron-silicate slag, which floats on top and which can be used as building material. A copper matte is also formed which is a mixture of copper, iron, and sulfur that is enriched in copper. The copper matte (30–70 % Cu) produced in the smelter is then introduced in a converter, where air is blown into the matte to remove the sulfur as SO2 gas and to form blister copper (>98 % Cu) and an iron-silicate “Fayalite” slag (Fe2SiO4). The blister copper is then put into an anode furnace (fire refining), where it is purified to anode-grade copper by removing most of the remaining sulfur, oxygen, and iron. Oxygen is usually removed by blowing natural gas through the melt (poling). The resulting anode copper (>99 % Cu) is further purified by electrorefining. The copper anodes are placed in an electrolysis set-up filled with an aqueous solution of CuSO4 and H2SO4. By applying a voltage, copper and the less noble metals (e.g., Fe, Ni, Co, Zn, Pb, Sb, Bi, As) dissolve at the anode, while the more noble metals (e.g., Ag, Au, Se, Te) settle at the bottom of the cell as anode slime. The Cu(II) ions migrate through the electrolyte to the cathode, and copper metal is deposited. Part of the less noble metal ions remain in solution as soluble sulfates (e.g., Fe, Ni, Co, Zn), while others precipitate as insoluble sulfates (Pb) or hydrolyzed species (As, Sb, Sn, Bi) and are therefore found in the anode slime on the bottom of the cell, together with the noble metals. The electrolyte solution and anode slimes are valuable by-products of copper electrorefining and can be further treated. Metals can also accumulate in the flue dust of the smelter or converter. A detailed overview of the existing copper production processes has been compiled by Moskalyk and Alfantazi [45]. Here, a condensed (schematic) overview of the copper production process and its residues is given (Fig. 6).

Schematic overview of primary copper production from copper ores (Color figure online)
The composition of the process residues is very dependent on the type of ore and the process that is used to extract the copper. However, some representative examples can be found in the literature (Table 4).
Antimony-containing residues are an increasingly big issue in copper processing due to the deteriorating quality of primary copper ores. However, no recycling activities are currently carried out on industrial scale although there is a strong commitment to develop these in the future. Antimony is found in different intermediates and residues of the copper production process, and a wide variety of processes has been developed on laboratory scale to recover antimony from these waste streams. Awe et al. [53] developed a flowsheet to remove antimony from copper concentrates which feed the copper industry. Alkaline sulfide leaching was used (Na2S/NaOH) to extract antimony from the ore, which was then electrodeposited as antimony metal. Using this approach, the antimony content in the concentrate was reduced from 1.7 % to less than 0.1 % Sb, which is desirable for copper. Zhang et al. [54] studied the recovery of antimony and bismuth from pressure-leached flue dust created during the smelting of copper. They used a three-step process consisting of a kerosene desulfurization step, a chloride leaching step, and a final hydrolysis step to recover 95.80 % of Bi and 90.8 % of Sb. Vircikova et al. [47] investigated the removal of arsenic from converter flue dust as well as the behavior of antimony and bismuth. Arsenic was leached using a Na2S solution and then precipitated using different methods. Recovery rates up to 99.9 % were achieved for arsenic but only 6.6 % for antimony. Fernández et al. [48] investigated the leaching of antimony and arsenic from anode slimes in the electrorefining of copper. The oxidized arsenic and antimony compounds in the anode slimes were selectively and almost completely dissolved in 0.4 M KOH at 80 °C. Meng et al. [49] studied the recovery of antimony(V) from the chloride leachate of copper anode slimes. Antimony was recovered by hydrolysis in the form of Sb2O5 with a recovery rate of 97 %. Anderson [7] reported the selective leaching of antimony and arsenic from copper electrorefining slimes, using alkaline sulfide leaching (Na2S–NaOH). Recovery rates were 99.3 % for Sb and 99.5 % for As. Li et al. [50] investigated alkaline fusion leaching as a method to recover valuable metals from copper anode slimes. The slime was fused with NaOH and NaNO3, followed by water leaching. Se, As, Sn, and Pb were leached out, leaving behind a valuable concentrate of Cu, Sb, Te, and precious metals (Ag, Au, Pt).
Much research has also been carried out to remove impurities (e.g., Sb, Bi, As, Pb) from copper electrolyte solutions with sorbents and solvent extraction methods [55]. Some metals such as antimony can cause passivation of the electrodes during electrorefining and is therefore important to remove these from the electrolyte solution [56]. Ando and Tsuchida [52] studied the recovery of trace impurities (Sb and Bi) from copper electrolyte solutions, using adsorbents with aminophosphonic acid functional groups. High recovery rates of 99.5 % for Bi and 100 % for Sb were achieved. Deorkar and Tavlarides [57] investigated the use of a silica gel ceramic support, functionalized with covalently bonded pyrogallol groups as a sorbent to separate antimony from copper and lead. The bed was regenerated by desorbing Sb(III) with HCl (4 M) and potassium hydrogen tartrate (0.05 M), resulting in a concentration factor of 25–30. Navarro and Alguacil [58] reported the adsorption of antimony and arsenic from a copper electrorefining solution onto activated carbon. Wang et al. [51] developed a new adsorbent based on Sb(V) and BaSO4 as a carrier to recover impurities (e.g., Sb, Bi) from a copper electrolyte solution. Sb(V) can combine with Bi(III) and Sb(III) to precipitate these ions as Bi(SbO4) and Sb(SbO4). The sorbent was able to remove 90 % of Bi and 80 % of Sb from the acidic copper electrolyte solution. Xiao et al. [59] conducted studies on the removal of antimony and bismuth impurities from synthetic copper electrolyte solution. They used As(III) ions as collector to precipitate the impurities, but low removal rates were reported: 53 and 52 % for Sb and Bi, respectively. Xiao et al. [60] also studied the removal of Sb, As and Bi impurities using Sb(III) ions as collector to precipitate these impurities from a synthetic copper electrolyte solution, but with low recovery rates: 48.0 % for Sb and 38.4 % for Bi.
Solvent extraction has also been studied as a way to purify the copper electrolyte solutions and to extract valuable metals such as antimony or bismuth [61]. Szymanowski [61] discussed the use of phosphor-based extractants (e.g., TBP, DEHPA, Cyanex® 923), hydroxamic acids, and (poly)alcohols as a way to recover Sb, As, and Bi from copper electrolytes. Navarro et al. [62] studied the extraction of antimony from copper electrorefining solutions with the hydroxamic acid extractant LIX 1104SM and HCl as a stripping agent. They found that the order of extraction was Sb(III) > As(V) > Fe(III) ≫ Cu(II), with a distribution factor D for Sb of 299. Sarkar and Dhadke [63] studied the solvent extraction separation of Sb(III) and Bi(III) with bis(2,4,4-trimethylpentyl) monothiophosphinic acid (Cyanex® 302) from H2SO4 and HCl solutions. They reported quantitative extraction of antimony and bismuth and separated the metals by first stripping with HNO3 (2 M) to remove bismuth and then H2SO4 (8.5 M) to strip antimony. Iyer and Dhadke [64] reported the quantitative extraction and separation of Sb(III) and Bi(III) from aqueous H2SO4 and HCl solutions with the trialkyl phosphine oxide extractant Cyanex® 925. Sb(III) was stripped from the organic phase with H2SO4 (8 M) and Bi(III) with HNO3 (2–3 M). Other relevant studies include the work of Fuyii et al. [65] on the extraction behavior of Sb(III) in the TRUEX® system and a study of the co-extraction of Sb(III) by the Cu(II) extractant Acorga® CLX50 performed by Lin [66]. This pyridine-3,5-dicarboxylate ester extractant is used in the CUPREX® process to produce cathode-grade copper with a hydrometallurgical process in which sulfide ore concentrates are leached in chloride medium [67]. The extraction of Sb(III) and Sb(V) ions with amine extractants in different acid solutions and the separation from other relevant metal ions (e.g., Bi(III), Sn(II), Cd(II), Te(IV), Se(IV), Pb(II), Cu(II), Au(III), Fe(III), and Zn(II)), have been studied extensively by Alian and Sanad [68] and Sargar et al. [69]. Facon et al. [70] also investigated the removal of Sb(III) from Bi(III), Pb(II), and Sn(IV) with Cyanex® 301 from chloride solutions.
Lead Production
Antimony is also connected to primary and secondary lead production because it occurs in primary lead ores (e.g., galena PbS) as well as in the lead alloys used in lead-acid (LA) batteries, which use up the majority of the world’s lead (76.7 % in 2006) [10]. Due to the large production volumes of lead (11,000,000 t/year in 2014), compared to antimony (160,000 t/year in 2014), lead production residues are considered as important secondary feedstocks of antimony [71]. Stringent environmental regulations and the exhaustion of high-quality primary lead deposits have caused an accelerated shift toward lead recycling as the main supply source [10]. In 2006, 60 % of the world’s annual lead consumption was met by recycling of LA batteries, lead pipes, etc. [10]. In the U.S.A., secondary supply meets more than 80 % of the demand for lead and in countries such as Austria, the Netherlands, Spain, and Belgium the entire lead production is based on secondary raw materials from lead recycling [72]. Here, an overview is given of primary and secondary lead production with an emphasis on the formation of the different Sb-containing residues (e.g., slags, drosses, speiss, slimes) and the existing technologies to recover antimony from these residues (Fig. 7). Almost all antimony in secondary lead is currently being recycled [73]. For example, antimonial drosses are used to produce antimonial lead [73]. Efforts have also started to upgrade the process in order to produce pure Sb2O3 from these secondary lead residues [74–76]. Residues with low antimony contents such as the slags and matte are less interesting when it comes to antimony recycling, but can have other applications [73]. Lead refiners are highly interested in valorizing the valuable Sb-containing waste streams both from an economic and environmental standpoint. The composition of the different process residues is dependent on the feed, equipment, and process, but some representative examples are shown in Table 5.

Schematic overview of primary lead production from lead ores, and secondary lead production from spent lead-acid (LA) batteries. The different metal-containing waste streams and residues are indicated in red [73] (Color figure online)
Primary lead production from lead sulfide ores (Galena: PbS) starts with the concentration of the ore, followed by a roasting and sintering step to form sinter (Fig. 7) [81, 82]. The lead concentrate is then fed into a blast furnace together with limestone (flux) and coke in order to reduce the oxides to the metal [81]. New reactor designs also allow the roasting and smelting in a single reactor (e.g., Isasmelt furnace) [2, 82]. The non-metallic fraction (e.g., sulfides, silicates) forms a slag with the fluxing materials. During the smelting, a lighter phase is formed which is known as speiss. Speiss consist mainly of iron arsenides and antimonides, and other elements such as Pb, Cu, Ni, Sn, and significant levels of precious metals (Ag 9 kg/t and Au 45 g/t) [7]. It rises to the top of the melt and can be skimmed off. Furthermore, a matte layer consisting of copper and other metal sulfide impurities is formed. The speiss and matte can be sold to copper smelters, where they are refined for copper recovery. The lead coming from the smelting furnace, called lead bullion, still contains many impurities (e.g., Cu, As, Sb, Sn, Bi, Zn, Ag, Au), and needs to be refined. First, copper is removed in a drossing process [81]. The melt is cooled to decrease the solubility of copper, and a copper dross is formed on the surface [81]. Sulfur can also be added to remove the last traces of copper. Dross is usually skimmed off and sent to a dross furnace to recover the non-lead components. The lead is then subjected to further purification steps either by thermal refining or by electrorefining [81]. In thermal refining, the lead is heated and cooled under different conditions and at different temperatures to oxidize or to remove the metal impurities from the lead. Arsenic, tin, and antimony are best removed in a softening step, which consists of heating the lead in a furnace in the presence of air, which causes the impurities with a greater affinity for oxygen than lead, to be oxidized. The softening skims can then be collected from the surface of the bath. An alternative is the Harris process, which uses a flux of molten NaOH and NaNO3 to remove the impurities as sodium arsenate, stannate, and antimonate [83, 84]. Further purification steps are used to remove gold and silver (Parkes process), zinc (vacuum distillation), bismuth (Kroll–Betterton process), and other impurities such as antimony (caustic refining) [2, 82, 84–86]. These refining processes are combined to obtain a lead bullion with sufficient purity, before casting it into ingots [81]. Instead of this sequence of thermal refining steps, pure lead can also be obtained by electrorefining after the initial decoppering step [87]. Solutions of H2SiF6 (Betts process) or HBF4 are used and the work-lead is cast into anodes and then dissolved [88]. The metals that are more noble than lead (e.g., As, Sb, Bi, Au, Ag) do not dissolve and are collected in the anode slimes [2]. The advantage of electrorefining is that unlike thermal refining all the impurities are removed in one step and 99.999 % pure lead is obtained [2]. In the sintering, smelting, and refining stages, fumes and dusts may contain between 10 and 60 % Pb and a wide range of other metals (e.g., Cu, Zn, As, Sb, Sn, Cd, Ag, Au, Bi). Plants are normally designed to capture these flue dusts in filters in order to discard them safely or resmelt them in certain cases [81].
Secondary lead production is almost entirely dependent on the recycling of spent lead-acid batteries (Fig. 7) [2, 73]. After removing the acid, the batteries are crushed to separate lead from the plastic. The lead can be separated in a lead metal fraction and a lead oxide fraction by a mechanical process [2]. The lead metal is then simply remelted, whereas the lead oxide is reduced in several types of furnaces (e.g., rotary furnaces or Isasmelt furnaces) [2, 11]. Besser et al. reviewed the different types of furnace designs and processes for battery recycling as well as their efficiency, and ability to produce pure lead and lead-antimony alloys for battery grid manufacturers and other lead alloy applications [10, 89]. The lead is refined using similar processes as the primary lead production and the same type of residues (e.g., skims, drosses, flue dust) are obtained [73].
The recovery of antimony from lead production residues has been investigated for some time [73]. Anderson [7], studied the use of alkaline sulfide leaching as a way to selectively remove antimony (and arsenic) from lead smelting residues. A mixture of Na2S and NaOH was used to solubilize antimony as thioantimonite Na3SbS3. High antimony recovery rates were reported for lead smelter speiss (99.4 %), skims (89.0 %), and flue dust (95 %) [7]. Singh [78] reported the recovery of antimony from the antimony-containing Harris dross (8.2 % Sb) of the refining section of a lead plant. HCl was used to leach antimony from the dross (95 % recovery) and Sb2O3 was formed by hydrolysis (73.6 % recovery) [78]. Peterson and Twidwell [90] investigated the removal of arsenic and antimony from lead smelter speiss, using volatilization techniques. The removal of antimony from antimony dust captured during the direct lead smelting process has been studied by Liu et al. They proposed a pyrometallurgical process based on reduction smelting, alkaline refining, and oxidation to extract antimony from antimony dust and produce Sb2O3 with a purity above 99.8 % [76]. Cao et al. studied the recovery of antimony from lead anode slimes, using a potential-controlled chlorination leaching ((Cl2) leaching) and continuous distillation [79]. High-purity SbCl3 was prepared, with an antimony recovery rate of more than 95 %. The process forms a closed loop and no waste is created [79]. Itoh et al. [91], proposed an alternative process to recover antimony from the anode slimes (Pb–Bi-Sb alloy) of spent lead-acid battery processing. In a method called volatile oxide formation, antimony is selectively oxidized due to its higher affinity for oxygen compared to lead or bismuth. Sb2O3 is then evaporated because of its high vapor pressure. Lin and Qiu [92] also developed a process to recover antimony and arsenic from anode slimes. They used a new process called vacuum dynamic flash reduction to evaporate the volatile oxides of antimony and arsenic, leaving silver behind. A recovery of 93.67 % was achieved for antimony and 98.92 % for arsenic. The vacuum treatment eliminates much of the air pollution and material losses associated with other conventional treatment methods. Qui et al. also used vacuum evaporation technology to recover antimony from Sb-rich anode slimes from the lead industry [93]. The antimony recovery rate was 92 %, and a 99.7 % pure antimony oxide distillate was obtained. Binz et al. [75] developed a bottom-up process to recover antimony from lead-refining residues. They investigated a more selective oxidation during lead softening and the carbothermic reduction of the slag to produce a concentrated antimony slag and improve the recovery of antimony. Kaporal et al. [94] studied the feasibility of antimony removal from accumulator acid found in spent lead-acid batteries. They achieved 100 % removal of antimony from synthetic solutions, using electrodeposition with a copper electrode. Bergmann and Kaporal [95] continued this work and studied the electrochemical recovery of antimony from real spent accumulator acid found in lead-acid batteries. They managed to reduce antimony levels from 5 ppm to less than 0.15 ppm, while also regenerating the accumulator acid. Copper and graphite cathodes produced the best results.
Spent Antimony Catalyst
Antimony is also used in several catalysts [96–101]. When these catalysts reach their end-of-life, antimony can be recovered and reused. The most important application is the production of polyethylene terephthalate (PET), one of the most common polyester thermoplastic polymers. This polymer is mostly used in textile fibers and as container for food or liquids. PET is produced by the Sb(III)-catalyzed polycondenzation of bis(hydroxyethyl)terephthalate (BHET) (Fig. 8). This precursor is either synthesized by the transesterification of dimethyl terephthalate (DMT) or the esterification of terephthalic acid (TPA). Titanium-based catalysts have been proposed, but today more than 90 % of the world’s polyester production still runs on Sb-based catalysts (150–300 ppm) [96, 102].

Two pathways for the production of PET (EG ethylene glycol)
The most common forms are Sb2O3, Sb(OCH2CH2O)3, and Sb(OAc)3 [103]. High-purity compounds are required, especially in regard to arsenic since the catalyst will be present in the final PET polymer used for making plastic bottles. Despite the efficiency of antimony catalysts, there are drawbacks such as the decomposition, which deposits antimony metal particles [103]. This causes a grayish discolouration in the polymer, especially at concentrations >250 ppm Sb [96]. The mechanism behind the formation of antimony metal during PET polymerization and the discolouration was studied in detail by Aharoni [104]. Although antimony is present in the PET polymer (100–300 ppm), it has been demonstrated that water stored in PET bottles contains less than 1 ppb of Sb, which is far below the EEC limit of 20 ppb [96, 105]. The mass recycling of PET plastic could therefore be an opportunity to recover antimony, although antimony contents are typically lower than in plastics containing antimony-based flame retardants. Therefore, most studies have focused on the recovery of antimony from spent catalysts so far. Dougherty and Garska [12] developed a process to recover antimony from spent ethylene glycol residues resulting from the manufacture of PET polymers. Their method consists of combusting the spent glycol residues to produce an ash, from which antimony can be recovered. Several patents have also been published on the recovery of antimony from halocarbon solutions, because Sb-containing catalysts (e.g., HF/SbCl5) are used in the fluorination of chlorinated hydrocarbons [99, 106, 107]. Fernschild et al. [106] described a method for the recovery of an Sb-containing catalyst by chlorination, followed by the distillation of SbCl5. Hyatt [107] developed an alternative process based on the reduction of SbCl5 to SbCl3, followed by extraction of SbCl3 from the halocarbon solution to an acidic aqueous phase. Finally, Anderson [7] also studied the recovery of antimony from a spent catalyst used in the production of acrolein from propylene. Antimony was successfully leached out (99.5 %) using HCl, leaving the silica substrate intact.
End-of-Life Products and Municipal Waste
Antimony can be found in a variety of products, which eventually end up as waste. It is therefore important to understand the presence of antimony in different items, in order to assure that no antimony is released into the environment. The high antimony content in certain products also makes them very interesting as secondary sources of antimony either through direct recycling or through municipal waste incineration. Table 6 gives an overview of the different antimony products and their approximate growth rates [3].
Municipal Solid Waste Incineration
Municipal solid waste incineration (MSWI) is a vital part of many waste management systems. The residues formed during the incineration contain many valuable elements and have been investigated as secondary source of ferrous and non-ferrous metals. In a MSWI plant, the waste is incinerated at a temperature of 850–1000 °C, forming a solid bottom ash fraction and flue gas, which is captured and cooled to form boiler ash, fly ash, and air pollution control (ACP) residues. A schematic overview of a MSWI plant is shown in Fig. 9.

Schematic representation of the MSWI plant in Doel, Belgium. This figure is reproduced with permission from KU Leuven SIM2 [108]
The waste incineration residues can undergo different treatments (e.g., dry, wet, thermal, mechanical) aimed at removing ferrous and non-ferrous metals and/or valorizing it as a granulate for the construction industry, e.g., in road foundation. A lot of different elements are present in these ashes, but the discussion is restricted here to the recovery of antimony. Jung et al. [109] and Allegrini et al. [13] compiled an overview of the composition of MSWI bottom ash, fly ashes, and other waste incineration residues, and compared this with typical ore concentrations to determine the recovery potential (Table 7) [13, 109]. The typical ore concentration given in Table 7 reflects the average grade of the deposits, but does not take into account the ore’s abundance. The widely available incinerator residues could therefore become an attractive secondary resource. Fly ashes are particularly interesting, due to the volatility of antimony which results in an important enrichment of antimony in the fly ashes. However, presorting of materials with high antimony contents (e.g., plastics with flame retardants), could drastically improve the efficiency of such an incineration process.
Nakamura et al. [111] investigated the origin of antimony, lead, and cadmium in incinerator waste. They concluded that the relatively high content of antimony in these incineration residues is mainly due to the presence of antimony in plastics, glass, and textiles (Table 8). Curtain fabrics had a particularly high antimony content (2100 ppm), due to the use of flame retardant coatings [111]. In general, Watanabe et al. [112] estimated that raw incinerator waste contains approximately 40–50 mg kg−1 of antimony.
The removal of antimony from incinerator residues has been an important topic of discussion, mainly because of its toxicity and the risk for uncontrolled release of antimony. Osaka et al. [113] wrote a report on the risks associated with the presence of antimony in municipal waste incineration residues. They studied the influence of the pH on the leaching of antimony from these residues and proposed risk management measures. Paoletti et al. (2001) and Nakamura et al. (1996) studied the movement and distribution of antimony in the different incinerator residues [111, 114]. These risk management studies focused mainly on limiting the leaching of antimony from these residues. However, due to the high criticality of antimony, the recovery of antimony from these residues may become of higher interest, especially since consumer waste contains important amounts of antimony in textiles, plastics, and glass (Table 8). Miravet et al. [115] analyzed the leachability and speciation of antimony in incinerator fly ash. They used citrate solutions to leach out antimony and concluded that Sb(V) was the main Sb-containing species. The extraction of metals from incinerator residues through leaching was also studied by Van Gerven et al. [110]. They performed column and batch leaching tests with acidified water (HNO3) to estimate the leachability of a wide range of metals from MSWI bottom ash, boiler ash, fly ashes, and air pollution control (APC) residues. Van Gerven et al. [116] also studied the removal of heavy metals from MSWI bottom ashes using organic solutions of citric acid and ammonium citrate. Cornelis et al. [14, 15] investigated the influence of the pH on the leaching of antimony from carbonated and non-carbonated MSWI bottom ash. Hong et al. studied the extraction of heavy metals from MSW incinerator fly ashes, using acids (HCl), and chelating agents (NTA, EDTA, DTPA) in batch experiments. The leaching test on the residues after the treatment with chelating agents showed that the fly ashes were successfully detoxified to meet the guideline for landfilling. Okkenhaug et al. [117] showed that iron-rich sulfuric acid waste is efficient to immobilize antimony in MSWI air pollution control residues. These technologies are all promising but the winning technology will be the one that can selectively remove certain elements such as antimony. Due to the mix of elements in these residues, it is crucial to develop methods which can deal with the complexity of these powders in a low-cost and efficient manner. Despite the technical challenges, there is a significant opportunity to recover valuable metals from these ashes as they are available in large quantities.
Flame Retardants in Plastics
In 2014, the world consumption of flame retardants was more than 2.2 million tons, representing a market of approximately 5 billion EUR with a 4–5 % annual growth rate due to rising safety standards worldwide [4, 118]. Flame retardants are crucial to guarantee the safety of plastics, construction materials, electronics, textiles, and coated products [119, 120]. There are three main classes of flame retardants: (1) metal hydroxides such as Al(OH)3 (ATH) and Mg(OH)2 (MDH), (2) organohalogen compounds (chlorinated or brominated) in combination with antimony trioxide (Sb2O3), and (3) organophosphorus compounds [119]. An overview of the flame retardant market (2007) is given in Fig. 10.

Flame retardant sales per region and per category (2007), expressed in t/year. Adapted from SRI-Consulting [121] (Color figure online)
Continuous research and regulations are driving new developments in the different categories of flame retardants. For example, research is being conducted to graft flame retardant groups in the structure of the polymer, thus avoiding the release of the flame retardants into the environment [122–124]. The most effective commercial fire retardant systems are currently based on brominated organic compounds (known as brominated flame retardants (BFRs)), preferably in conjunction with antimony trioxide Sb2O3 due to the strong synergetic effect [120]. Flame retardants were responsible for 52 % of the demand for antimony in 2010 (≈103,500 t) [3]. Camino and Costa [119], reviewed the performance and mechanisms of antimony fire retardants in polymers. They attributed the synergetic effect to the fact that metal halides are far more effective flame inhibitors, compared to the hydrogen halides formed in the absence of the metal compound [120]. In reality, a variety of different oxyhalide metal species are formed and their thermal stability and chemical reactivity have therefore been studied in great detail [125]. A discussion of the fire retardant mechanism is beyond the scope of this work, and what follows is therefore only a discussion of the possible recovery of antimony from plastics, electronics, and coatings. The antimony content in plastics is highly dependent on the type of plastic and the application. Table 9 provides an overview of the typical formulations used to fireproof different polymers [6].
Non-halogenated polymers are inherently flammable and require large amounts of halogenated flame retardants in combination with Sb2O3 to be flame resistant [6]. Halogenated polymers (e.g., PVC, PVDC) on the other hand, are already partially flame retarded due to their chlorine content and do not require the addition of halogenated compounds. Smaller amounts of Sb2O3 can be added to these polymers to ensure their fire resistance [6]. Due to the inherent risk for fires, polymers used in building materials, electronics, and some textiles are often made fireproof using these halogenated compounds and Sb2O3. Plastics used for food and beverages or plastics that do not require fire-proofing, do not contain significant quantities of antimony. PET bottles are an exception, as these contain some antimony (0.2–0.3 wt%) due to the use of an antimony catalyst for the production of PET polymers [126]. Efficient recovery of antimony from plastics requires adapted screening and sorting methods to identify plastics with high antimony contents. X-ray fluorescence spectroscopy (XRF) could provide a fast and non-destructive screening. Other interesting techniques are X-ray transparency and laser-induced breakdown spectroscopy (LIBS). Bellara et al. [127] also developed a method for the direct determination of antimony in solid PVC samples by graphite furnace atomic absorption spectrometry (GFAAS). They concluded that this screening technique provides sufficiently reliable results in a reasonably short time, but it is difficult to implement in a continuous process. Once sorted, the antimony-containing plastics can be placed in special ovens for the pyrolysis of the polymer, where antimony is caught in the residues (bottom ashes or fly ashes). A patent of Nippon Electric Co (1994) described a method to decompose a resin, containing flame retardants, and capture the bromine and antimony in a gas mixture (e.g., HBr, SbBr3, CO2, H2O) [128]. By washing the gas mixture with an alkaline aqueous solution (e.g., NaOH), NaBr and Sb2O3 could be obtained. The thermal decomposition could be operated under air-free conditions or by combustion. In a follow-up patent, Masatochi et al. [129] specifically described the recovery of antimony from decomposition gasses, originating from the pyrolysis of flame resistant plastics. Masatoshi and Ikuta [130] also described the pyrolysis-based recovery of antimony and bromine from molding resins used in electronics. The electronic parts consisted of an epoxy resin, a silica filler, and flame retardants consisting of brominated compounds and Sb2O3. This study investigated the best pyrolysis conditions to obtain a pure silica product. The exhaust gasses generated in the pyrolysis of the molding resin were treated to remove the bromine and convert the antimony bromides to Sb2O3. Jakab et al. [131] studied the thermal decomposition of flame-retarded high-impact polystyrene. In a detailed overview of the decomposition pathways and mechanisms, they concluded that the brominated additives themselves do not change the decomposition temperature of polystyrene, but Sb2O3 reduces the thermal stability of the samples by acting as an initiator for the decomposition of the brominated flame retardants. Klein et al. [132] also studied the behavior of antimony during thermal treatment of antimony-rich halogenated waste. Their aim was to ensure the environmentally friendly waste incineration of antimony-rich plastic waste, by strengthening the knowledge about the fate of antimony and the potential formation of harmful species as function of temperature, residence time and flow rate of air in the furnace. Their investigations showed that under moderate oxidative conditions, the partition of antimony between the residual bottom ash (≈64 %) and the gas phase (≈36 %) was constant regardless of the temperature. However, they observed that at 850 °C antimony was mainly present in the gas phase as Sb(III), while around 1100 °C, Sb(V) was favored. Wu et al. [133] studied the thermal degradation of high-impact polystyrene, containing brominated flame retardants and Sb2O3. Bauxite residue (red mud) was used as an additive to catch the bromine and antimony in a residue, while producing a valuable pyrolysis oil. Mitan et al. [134] investigated the controlled pyrolysis of polyethylene/polypropylene/polystyrene mixed with brominated high-impact polystyrene. Their work describes the effect of the brominated flame retardants and Sb2O3 on the pyrolysis process and the distribution of pyrolysis products. Brebu et al. [135] also looked at the effect of flame retardants and Sb2O3 synergists on the thermal decomposition of high-impact polystyrene and on its debromination by ammonia treatment. More than 90 wt% of initial bromine could be recovered as inorganic NH4Br. Hall et al. [136] developed a method for the co-pyrolysis of flame-retarded high-impact polystyrene and polyolefins in a fixed bed reactor. They looked at the effect that the different types of brominated aryl compounds and Sb2O3 have on the pyrolysis products.
Besides pyrolysis, other methods have also been proposed. Omwudili and Williams [137] attempted the alkaline reforming of high-impact polystyrene and traced the movement of bromine and antimony. The waste polymer was reacted in supercritical water and NaOH in a pressure reactor in order to neutralize the corrosive inorganic bromine species released during the reactions. They observed that the presence of the alkaline supercritical water led to 97 wt% debromination of the product oil, producing a valuable bromine-free oil feedstock. Furthermore, a 98 % removal rate was achieved for antimony. The same method was also used for acrylonitrile–butadiene–styrene polymers [138]. Another unusual approach was tested by Lateef et al. [139]. They used imidazolium- and pyridinium-based ionic liquids to extract brominated flame retardants and Sb2O3 from high-impact polystyrene. They identified 1-hexylpyridinium bromide as the most suitable for solid–liquid and liquid–liquid extraction, achieving extraction percentages of 92.7 % for the brominated flame retardants and 99.9 % for Sb2O3. Furthermore, they demonstrated that the chain length of the polymer was not degraded by the extraction process, thus forming a polymer residue which can be recycled. Another approach could be the depolymerization of the polymers to recover Sb2O3 and the building blocks (e.g., ethylene, HCl). This approach has not been investigated in detail so far, but some projects have looked into the possibility of separating, cleaning, and reusing bromine- and antimony-containing plastics [140].
Plastics in Waste Electrical and Electronic Equipment
A subcategory of flame retardant plastics are the plastics used in waste electrical and electronic equipment (WEEE) [141]. WEEE is already being recycled on a large scale, mainly to recover the precious metals (e.g., Co, In, Te, Se, Au, Ag) used in the electronic components such as printed circuit boards (PCBs) (Table 10) [142]. Besides the metallic fraction, there is also a large non-metallic fraction in WEEE [143]. Even for PCBs the non-metallic fraction is still more than 70 % [143]. This non-metallic fraction includes the silicon substrates and the plastics used in these products. The strict safety regulations for electrical and electronic equipment (EEE), require large amounts of brominated flame retardants and Sb2O3 to be used in order to guarantee the safe use of these plastics [142]. As explained previously, approximately 52 % of antimony is consumed in flame retardants, of which 70 % is used for plastics in electrical and electronic equipment (Table 6) [3, 142].
This clearly shows the potential of WEEE plastics as a secondary feedstock of antimony. Table 9 showed that certain plastics require as much as 5–15 wt% of antimony to be fire resistant, making WEEE particularly rich in antimony. The most important plastics under investigation are acrylonitrile–butadiene–styrene (ABS) and high-impact polystyrene (HIPS) as these are the most-used polymers in EEE (Fig. 11) [141]. These polymers typically contain 5–10 wt% of Sb2O3 and an even larger amount of brominated flame retardants. [142]. Different studies have been carried out to deal with these polymers in WEEE recycling schemes. The toxic brominated compounds and antimony-containing plastics have been a source of concern for the recycling of WEEE. Regulations for the emission of brominated compounds are very strict and therefore solutions have been investigated to capture the bromine efficiently during the pyrolysis of the WEEE [142]. The fact that antimony is also present in the plastic fraction, means that bromine and antimony are intrinsically connected in WEEE recycling.

Flows of the major plastic types in Swiss WEEE products for the year 2007 [141]. ABS acrylonitrile–butadiene–styrene, HIPS high-impact polystyrene, PP polypropylene, PC polycarbonate, PU polyurethane, VC polyvinylchloride
De Marco et al. and Moltó et al. reviewed the pyrolysis of electrical and electronic wastes but did not discuss in detail the recovery of antimony or bromine flame retardants [144, 145]. Guo et al. [143] reviewed the recycling of non-metallic fractions (NMFs) from waste printed circuit boards (PCBs) and demonstrated the importance of finding a solution for the recycling of this important fraction of WEEE. The non-metallic fraction was usually treated by combustion or landfilling in the past. However, uncontrolled combustion causes the formation of highly toxic polybrominated dibenzodioxins and dibenzofurans, while landfilling of the NMFs will lead to secondary pollution caused by heavy metals and brominated flame retardants (BFRs) leaching to the groundwater. This had been previously demonstrated by the work of Gullet et al. [146], who characterized the emissions and residual ash from open air burning of electronic wastes during simulated rudimentary recycling operations. They analyzed the fly ash and particulate matter and characterized the organic and metallic components in the air emissions. Therefore, Guo et al. [143] concluded that more advanced processes are required such as pyrolysis, gasification, depolymerization, or hydrogen degradation in order to convert the non-metallic fraction to chemical feedstocks and fuels. These methods have the advantage of eliminating hazardous substances such as brominated organic compounds from this waste fraction and allow the recovery of antimony from the plastic waste. In later work, Guo et al. [147] analyzed the pyrolysis of scrap printed circuit board plastic particles in a fluidized bed reactor. The gas products, liquid products, and solid residues were analyzed and it was found that the liquid yields increased with an increase in pyrolysis temperature. The main compositions of liquid products were aromatic compounds including substituted benzenes, whereas the solid products mainly contained char and fiberglass. Morf et al. [148] also tracked the movement of brominated flame retardants and antimony in WEEE recycling plants. Hall et al. [149] investigated the fast pyrolysis of plastics recovered from computer waste. The computer case was made from PVC, but the computer monitor cover consisted of brominated acrylonitrile–butadiene–styrene and had an antimony content between 3.9 and 4.5 wt%. The pyrolysis was carried out in a fluidized bed reactor at 500 °C, and the composition products were studied. They concluded that the high bromine and chlorine content was a nuisance and that in-process sorption of the halogens was required to obtain a high-quality oil. In later work, Hall et al. [150] used zeolite catalysts and fluidized catalytic cracker (FCC) catalysts to destroy the toxic organobromines during pyrolysis of brominated high-impact polystyrene (4.6 wt% Sb) and acrylonitrile–butadiene–styrene (3.2 wt% Sb), found in WEEE. The movement of antimony in the oil and char residues was also tracked. Tostar et al. [151] specifically investigated the leaching of antimony in plastics from WEEE with various acids and gamma irradiation. The most efficient leaching medium was a heated solution of sodium hydrogen tartrate in dimethyl sulphoxide, which leached approximately 50 % of the antimony from the acrylonitrile–butadiene–styrene plastic. Gamma irradiation did not significantly change the amount of leached antimony. A patent by Zhenming et al. [152] also describes vacuum distillation as a way to recover antimony from printed circuit board waste. Yang et al. [153] reviewed the pyrolysis and dehalogenation of plastics from waste electrical and electronic equipment. The conclusion from these works is that there are technologies available to valorize the plastic components of WEEE, but that additional research needs to be done to improve the efficiency of these processes before upscaling becomes economically feasible. These plastics have to be processed appropriately due to their high content of toxic brominated compounds. The focus up till now has been on limiting the emission of brominated compounds and producing useful fuel oils. However, the high antimony content present in these same plastics offers a unique opportunity to combine the necessary treatment of these brominated plastics with the recovery of valuable antimony. Valorizing antimony from WEEE plastics could improve the economics of the total WEEE recycling process and offset the additional cost of de-brominating during the processing of WEEE plastics.
Lamp Phosphor Waste
Antimony is used in the halophosphate (HALO) lamp phosphors, found in fluorescent lamps [154, 155]. These lamp phosphors are coated on the inside of the glass and are responsible for the emission of visible light [156–159]. The HALO phosphor was invented in 1942 and large amounts of this phosphor were produced as it found widespread use in fluorescent tubes due to its emission of intense white light [156–159]. This phosphor usually consists of a fluoro-chloro apatite doped with Mn(II) and Sb(III) and has the general formula (Sr,Ca)5(PO4)3(Cl,F):Sb3+, Mn2+ [156–158]. Antimony is added as a blue dopant to adjust the shade of white [156–158, 160]. Today, lamp phosphor waste contains around 50 wt% of HALO, making it the largest component [154, 155, 161]. This corresponds to approximately 0.5–1 wt% of antimony which is similar to a low-grade stibnite ore [1, 155, 157, 161, 162]. With annual sales of CFLs topping 2.5 × 109 units/y (2007), the volume of lamp phosphor waste is expected to continue growing at a rapid pace as fluorescent lamps reach their end-of-life [163, 164]. Due to their mercury content, fluorescent lamps are considered as hazardous waste and are therefore collected separately in most countries [165, 166]. The interest in lamp phosphor powders as a secondary resource has been growing recently due to their high rare-earth content (in tri-band phosphor), and the recovery of these valuable elements has been studied extensively [165–175]. Currently, a recycling process is operated on industrial scale (>1000 t/y) by Solvay in France [161]. So far, the Sb-containing HALO phosphor is often still discarded as a non-valuable residue due to the absence of rare earths [165, 166, 172]. However, the HALO content in these powders is too high to ignore (50 wt%) and could serve as an interesting secondary source of antimony [176]. Our group therefore recently developed a zero-waste valorization method to recover antimony from HALO and to valorize the remaining as a calcium phosphate (apatite) product, which is a feedstock for the fertilizer industry (Fig. 12) [176]. The valorization of HALO and the recovery of antimony can be integrated in rare-earth recovery schemes and in the broader effort to recycle these lamp phosphor powders [175, 176].

Zero-waste valorization process for the recovery of antimony from the halophosphate fraction of lamp phosphor waste. The phosphor is first dissolved in HCl, followed by the selective extraction of Sb(III) with Aliquat® 336. Pure Sb2O3 is then obtained after a scrubbing (NaCl) and stripping step (NaOH). The remaining leachate is precipitated as apatite (fertilizer precursor). Reproduced with permission from Ref. [176]. Copyright Royal Society of Chemistry (2016) [176]
Antimony Emissions and Recovery
Antimony Emissions: Land, Water, Air
Antimony is also lost through emissions in the air, in the water, and on the land [177]. Total antimony emissions are difficult to estimate, but it has been shown that in Australia alone, total emissions of antimony (air, water, land) reach as much as 3.8 t/year during mining and 8.7 t/year during metal manufacturing. A better understanding of these losses can help limit the unwanted emissions of antimony and increase the available supply of antimony at the same time. Land emissions are important, especially around antimony mining and processing sites. An overview of antimony emissions in the soil and decontamination procedures has been compiled by Wilson et al. [178] and since this antimony is often very localized and difficult to access, it is not discussed here in detail. Air emissions on the other hand are very relevant to the topic of secondary sources of antimony. We previously discussed how air pollution control residues, flue dusts, and fly ashes in lead, copper, gold, and antimony processing can contain important amounts of antimony. An overview was also given of the different valorization methods and antimony recovery processes for these residues. However, the capture of antimony air emissions still needs to be improved. Tian et al. [177] made a study of global yearly air emissions and calculated that in 2005 global air emissions of antimony reached 2232 t, and then gradually declined to about 1904 t in 2010. Atmospheric emissions are mostly caused by fuel combustion (42 %), waste incineration (24 %), brake wear (17 %), and metal production (17 %) [177]. China is the largest emitter of atmospheric antimony (649 t in 2010) due to the large-scale combustion of coal and the production of metals, as China currently has 114 antimony mines dispersed over 18 provinces [177]. The emission of antimony from coal power plants, waste incinerators, and metal production sites, can be lowered by the implementation of better filters and air pollution control systems, from which the antimony can then be recovered and reused. However, the emissions of antimony through the wear of vehicle brake linings and brake pads has proven to be a larger concern as antimony emissions are not localized and no simple solutions are available. Sb2S3 is used as a lubricant in disc brake pads and drum brake pads, with concentrations as high as 46 g/kg, depending on the type of brake [179, 180]. Uexküll et al. [180] and Varrica et al. [179] studied the speciation of these brake wear particles and concluded that while the brake linings are mainly composed of Sb2S3, this compound is decomposed during the abrasion process leading to a mix of Sb2O3 and Sb2O5 in the airborne particulate. Finally, there is the emission of antimony to surface water, which has been reviewed by Filella et al. [181]. These emissions can also be important secondary sources of antimony if proper filtration systems are set in place. In concentrated industrial flows and mine drainage this is simple to implement, but even for very dilute water streams, decontamination is mandatory, so these large volumes can result overall in a significant amount of recovered antimony.
Antimony Removal from Waste Water
Antimony removal from waste water was recently reviewed by Ungureanu et al. [182]. This extensive review gives an overview of the analytic techniques for antimony speciation and the existing removal techniques [182]. Different removal techniques have been proposed such as ion-exchange, adsorption, membrane separation, electrochemical methods, bio-removal methods, or coagulation/flocculation followed by filtration or sedimentation [182]. The very large number of papers written on this topic, clearly demonstrated the importance of antimony removal from waste water [182]. In these studies, the focus is systematically on the removal of antimony to obtain clean water. This field of study is different from the previous chapters in this review, as it deals with very dilute antimony waste streams. However, the large volumes represent a sizeable amount of antimony, which should not go to waste. Antimony removal from wastewater is therefore an opportunity to collect antimony efficiently, using automatic (continuous) technologies such as column filtration with specialized sorbents. These techniques lead to a strong concentration of the antimony, which can then be used as a source of antimony, when renewing or regenerating the sorbent or filter.
The most common treatment techniques for antimony removal are flocculation–coagulation followed by filtration or sedimentation, or the use of adsorbents [182]. Flocculation–coagulation is one of the most employed treatments to produce drinking water, and has been investigated for antimony removal as well, with ferric chloride being the most suitable coagulant [182–185]. The drawback of this technique is that it is difficult to automate, it creates toxic sludge and it is not 100 % effective at removing antimony. Researchers have therefore increasingly turned to adsorbents due to the convenient automatization in a continuous set-up and the generally higher antimony-removal efficiencies [182]. The use of sorbents requires only a periodic regeneration, which is an opportunity to recover (pure) antimony. A large variety of sorbents have been investigated to this effect, and a full list is not included here since these have already been reviewed by Ungureanu et al. [182]. The investigated materials include chars, activated carbons, graphene, alumina, metal oxides, minerals, resins, biosorbents, and sorbents based on waste materials and residues. The benchmark was set by Xu et al. [186] who achieved the highest adsorption capacity (214 mg/g) using a binary iron-manganese oxide, whose adsorption capacity significantly outperformed the individual oxides [182, 186]. However, it should be stated that this result is an outlier, with most adsorbents having an adsorption capacity around 50–100 mg/g [182]. The second best material was prepared by Biswas et al. [187], who used saponified orange waste, loaded with Zr(IV) and Fe(III), as a sorbent for the adsorptive removal of Sb(III) and Sb(V) from waste water. They reported adsorption capacities around 114–145 mg/g. However, the review by Ungureanu et al. [182] does not yet include the latest results and an overview of some additional recent examples is given here, which boast high adsorption capacities (>100 mg/g). Wang et al. prepared iron-modified aerobic granules to remove Sb(V) from waste water and investigated the adsorption mechanism in detail [188]. They reported a maximal adsorption capacity of 125 mg/g. Dou et al. [189] investigated hierarchical macro- and mesoporous amorphous alumina as a sorbent for Sb(V). The results were compared with five different commercial alumina adsorbents and showed that this material had the highest adsorption capacity (118 mg/g) and fast adsorption kinetics. Dong et al. [190] made nanocomposites from graphene oxide and schwertmannite (iron oxyhydroxysulphate mineral) to adsorb Sb(V) from aqueous solutions. A synergetic effect on Sb(V) uptake was observed for this composite adsorbent, which had a maximum Sb(V) adsorption capacity of 158.6 mg/g. Sb(V) in spiked tap water (100 μg/L), simulated river water (6400 μg/L), and acid mine drainage (50,000 μg/L) was adsorbed by this sorbent to levels well below the regulation levels for these waters. An interesting subcategory of water purification is the removal of antimony from mine drainage. These flows are often much more concentrated and more acidic and therefore demand different solutions. Zhu et al. used electrocoagulation to partially remove antimony from mine flotation waste water [191]. The remaining concentration was still 1 mg/L, which far exceeds the drinking water limit, but meets the emission standards established by the state department of environmental protection of China [191].
Conclusion and Outlook
Antimony is one of the most critical elements, but has remained largely out of the spotlights. However, the gap between supply and demand is expected to exceed 10 % in the coming years, making antimony supply far more critical than for example the rare-earth elements. Similar to the rare earths, antimony production is dominated by China and as the grades of primary ores diminishes, it becomes increasingly difficult to meet the demand for antimony. Antimony is an important industrial commodity and is used in flame retardants for plastics and electronics, but also in catalysts for the production of PET polymers and in lead-acid batteries. To meet demand, it is crucial that attention shifts to secondary sources such as industrial process residues and end-of-life consumer items. This review gives an overview of existing waste streams which could be of importance as secondary sources of antimony. It also encourages researchers to look at unusual sources such as air pollution control residues, low-grade ores, and unusual products such as lamp phosphor waste. Sufficient technologies are available to make antimony recycling a success and current recycling rates of approximately 20 % are expected to rise as China struggles to increase its output. However, one of the remaining obstacles for the implementation of antimony recycling in the industry is the upscaling from laboratory methods to industrial processes. Further work needs to be carried out on pilot scale to see which methods are sufficiently robust and flexible. Another challenge is the changing composition of various residues depending on the ore or product that is being processed. Changes in composition of the input material can greatly affect the performance of the recovery processes. Finally, economic feasibility studies and life cycle assessments need to be carried out to determine which techniques are the most promising for industrial upscaling. We expect copper producers to invest in antimony recovery techniques in coming years as this is a growing problem that needs to be addressed in the copper production flowsheets. Other very interesting sources include incineration ashes from concentrated fractions such as textiles or flame retardant plastics. Recycling of antimony through the recycling of lead-acid batteries is currently carried on industrial scale and should continue to provide a steady stream of secondary antimony. Current primary antimony production is unsustainable as current reserves account for only 10–11 years of production at current levels which could be aggravated by rising demand from existing and future antimony applications such as liquid–metal batteries and thermoelectric materials. Moreover, antimony is also associated with environmental problems, so a better management of antimony resources is mandatory and the antimony life cycle must become more circular in the coming years.
References
Anderson CG (2012) The metallurgy of antimony. Chem Erde 72:3–8. doi:10.1016/j.chemer.2012.04.001
Claude F (2002) Non-ferrous metal: from Ag to Zn. Lannoo, Tielt
Chegwidden J (2011) Study of the antimony market. Roskill Consulting Group Ltd, London
Satterthwaite K (2014) Flame retardant markets to 2018. Roskill Consulting Industrial Minerals Event, Budapest
Schmidt M, Liedke M, Domer U (2015) Rohstoffrisikobewertung—Antimon. Deutsche Rohstoffagentur (DERA) in der Bundesanstalt für Geowissenschaften und Rohstoffe (BGR), Berlin, Germany
Uses and Formulations. (2015) United States Antimony Corportation. http://www.usantimony.com/2013_uses_formulations.htm
Anderson CG (2001) Hydrometallurgically treating antimony-bearing industrial wastes. JOM 53:18–20. doi:10.1007/s11837-001-0156-y
Antimony Statistics and Information (2014) U.S. Geological Survey (USGS), USA
Report on Critical raw materials for the EU (2014) European Commission, DG Enterprise & Industry, Brussels
Besser AD, Sorokina VS, Sokolov OK, Paretskii VM (2009) Processing of utilized lead-acid storage batteries—the basis of lead recycling. Russ Metall 2009:781–787. doi:10.1134/S0036029509080217
Ramus K, Hawkins P (1993) Lead/acid battery recycling and the new Isasmelt process. J Power Sources 42:299–313. doi:10.1016/0378-7753(93)80159-M
Dougherty SJ, Garska KJ (1978) Recovery of sodium and antimony values from spent ethylene glycol residues. U.S. Patent US4100253 A
Allegrini E, Maresca A, Olsson ME, Holtze MS, Boldrin A, Astrup TF (2014) Quantification of the resource recovery potential of municipal solid waste incineration bottom ashes. Waste Manage 34:1627–1636. doi:10.1016/j.wasman.2014.05.003
Cornelis G, Gerven TV, Vandecasteele C (2012) Antimony leaching from MSWI bottom ash: modelling of the effect of pH and carbonation. Waste Manage 32:278–286. doi:10.1016/j.wasman.2011.09.018
Cornelis G, Van Gerven T, Vandecasteele C (2006) Antimony leaching from uncarbonated and carbonated MSWI bottom ash. J Hazard Mater 137:1284–1292. doi:10.1016/j.jhazmat.2006.04.048
Yang J-G, Wu Y-T (2014) A hydrometallurgical process for the separation and recovery of antimony. Hydrometallurgy 143:68–74. doi:10.1016/j.hydromet.2014.01.002
Macdonald MD, Stevens DA, Thibault JD (1997) Process for producing antimony trioxide. WO 1998011021 A1
Guo X, Wang K, He M, Liu Z, Yang H, Li S (2014) Antimony smelting process generating solid wastes and dust: characterization and leaching behaviors. J Environ Sci 26:1549–1556. doi:10.1016/j.jes.2014.05.022
Fuxu H, Hongjiu R, Zhe S, Qingwen L (1989) Three-phase fluidized bed leaching of antimony refining slag. Hydrometallurgy 23:119–125. doi:10.1016/0304-386X(89)90022-4
Luo H-L, Liu W, Qin W-Q, Zheng Y-X, Yang K (2016) Cleaning of high antimony smelting slag from an oxygen-enriched bottom-blown by direct reduction. Rare Metals. doi:10.1007/s12598-015-0468-7
Keqiang Q, Rongliang Z (2006) Research on preparation of nanometer antimony trioxide from slag containing antimony by vacuum evaporation method. Vacuum 80:1016–1020. doi:10.1016/j.vacuum.2006.01.010
Salihoglu G (2014) Immobilization of antimony waste slag by applying geopolymerization and stabilization/solidification technologies. J Air Waste Manag Assoc 64:1288–1298. doi:10.1080/10962247.2014.943352
Courtin-Nomade A, Rakotoarisoa O, Bril H, Grybos M, Forestier L, Foucher F, Kunz M (2012) Weathering of Sb-rich mining and smelting residues: insight in solid speciation and soil bacteria toxicity. Chem Erde 72:29–39. doi:10.1016/j.chemer.2012.02.004
Anawar H, Freitas MC, Canha N, Santa Regina I (2011) Arsenic, antimony, and other trace element contamination in a mine tailings affected area and uptake by tolerant plant species. Environ Geochem Health 33:353–362. doi:10.1007/s10653-011-9378-2
Yang J-G, Tang C-B, Chen Y-M, Tang M-T (2011) Separation of antimony from a stibnite concentrate through a low-temperature smelting process to eliminate SO2 emission. Metall Mater Trans B 42:30–36. doi:10.1007/s11663-010-9453-6
Ye L, Tang C, Chen Y, Yang S, Yang J, Zhang W (2015) One-step extraction of antimony from low-grade stibnite in sodium carbonate—sodium chloride binary molten salt. J Clean Prod 93:134–139. doi:10.1016/j.jclepro.2015.01.018
Gök Ö (2014) Catalytic production of antimonate through alkaline leaching of stibnite concentrate. Hydrometallurgy 149:23–30. doi:10.1016/j.hydromet.2014.06.007
Roper AJ, Williams PA, Filella M (2012) Secondary antimony minerals: phases that control the dispersion of antimony in the supergene zone. Chem Erde 72:9–14. doi:10.1016/j.chemer.2012.01.005
Baláž P, Achimovičová M (2006) Selective leaching of antimony and arsenic from mechanically activated tetrahedrite, jamesonite and enargite. Int J Miner Process 81:44–50. doi:10.1016/j.minpro.2006.06.004
Awe SA, Sandström Å (2010) Selective leaching of arsenic and antimony from a tetrahedrite rich complex sulphide concentrate using alkaline sulphide solution. Miner Eng 23:1227–1236. doi:10.1016/j.mineng.2010.08.018
Yang T-Z, Jiang M-X, Lain Q-L, Chen J-Z (2005) Sodium sulfide leaching of low-grade jamesonite concentrate in production of sodium pyroantimonate. J Cent South Univ Technol 12:290–294
Duchao Z, Qingkai X, Weifeng L, Lin C, Tianzu Y, Younian L (2015) Pressure oxidation of sodium thioantimonite solution to prepare sodium pyroantimonate. Hydrometallurgy 151:91–97. doi:10.1016/j.hydromet.2014.11.014
Kanarskii AV, Adamov EV, Krylova LN (2012) Flotation concentration of the sulfide antimony-arsenic gold-bearing ore. Russ J Non-Ferr Met 53:120–124. doi:10.3103/S1067821212020058
Solozhenkin PM, Alekseev AN (2010) Innovative processing and hydrometallurgical treatment methods for complex antimony ores and concentrates. Part I. J Miner Sci 46:203–209. doi:10.1007/s10913-010-0026-5
Celep O, Alp İ, Deveci H (2011) Improved gold and silver extraction from a refractory antimony ore by pretreatment with alkaline sulphide leach. Hydrometallurgy 105:234–239. doi:10.1016/j.hydromet.2010.10.005
Karimi P, Abdollahi H, Amini A, Noaparast M, Shafaei SZ, Habashi F (2010) Cyanidation of gold ores containing copper, silver, lead, arsenic and antimony. Int J Miner Process 95:68–77. doi:10.1016/j.minpro.2010.03.002
Saleh SM, Said SA, El-Shahawi MS (2001) Extraction and recovery of Au, Sb and Sn from electrorefined solid waste. Anal Chim Acta 436:69–77. doi:10.1016/S0003-2670(01)00866-2
Kyle JH, Breuer PL, Bunney KG, Pleysier R (2012) Review of trace toxic elements (Pb, Cd, Hg, As, Sb, Bi, Se, Te) and their deportment in gold processing: part II: deportment in gold ore processing by cyanidation. Hydrometallurgy 111–112:10–21. doi:10.1016/j.hydromet.2011.09.005
Kyle JH, Breuer PL, Bunney KG, Pleysier R, May PM (2011) Review of trace toxic elements (Pb, Cd, Hg, As, Sb, Bi, Se, Te) and their deportment in gold processing. Part 1: mineralogy, aqueous chemistry and toxicity. Hydrometallurgy 107:91–100. doi:10.1016/j.hydromet.2011.01.010
Ubaldini S, Vegliò F, Fornari P, Abbruzzese C (2000) Process flow-sheet for gold and antimony recovery from stibnite. Hydrometallurgy 57:187–199. doi:10.1016/S0304-386X(00)00107-9
Milham L, Craw D (2009) Antimony mobilization through two contrasting gold ore processing systems, New Zealand. Mine Water Environ 28:136–145. doi:10.1007/s10230-009-0071-y
Celep O, Alp İ, Paktunç D, Thibault Y (2011) Implementation of sodium hydroxide pretreatment for refractory antimonial gold and silver ores. Hydrometallurgy 108:109–114. doi:10.1016/j.hydromet.2011.03.005
Solozhenkin PM, Alekseev AN (2010) Innovative processing and hydrometallurgical treatment methods for complex antimony ores and concentrates. Part II: hydrometallurgy of complex antimony ores. J Min Sci 46:446–452. doi:10.1007/s10913-010-0056-z
Korhonen JM, Välikangas L (2014) Handbook of organizational and entrepreneurial ingenuity. Edward Elgar Publishing Inc, Cheltenham, U.K
Moskalyk RR, Alfantazi AM (2003) Review of copper pyrometallurgical practice: today and tomorrow. Miner Eng 16:893–919. doi:10.1016/j.mineng.2003.08.002
Zivković Ž, Mitevska N, Mihajlović I, Nikolić Đ (2009) The influence of the silicate slag composition on copper losses during smelting of the sulfide concentrates. J Min Metall Sect B 45:23–24. doi:10.2298/JMMB0901023Z
Vircikova E, Havlik M (1999) Removing as from converter dust by a hydrometallurgical method. JOM 51(9):20–23. doi:10.1007/s11837-999-0152-1
Fernández MA, Segarra M, Espiell F (1996) Selective leaching of arsenic and antimony contained in the anode slimes from copper refining. Hydrometallurgy 41:255–267. doi:10.1016/0304-386X(95)00061-K
Meng L, Zhang S-G, Pan D-A, Li B, Tian J-J, Volinsky AA (2015) Antimony recovery from SbCl5 acid solution by hydrolysis and aging. Rare Met 34:436–439. doi:10.1007/s12598-015-0480-y
Li D, Guo X, Xu Z, Tian Q, Feng Q (2015) Leaching behavior of metals from copper anode slime using an alkali fusion-leaching process. Hydrometallurgy 157:9–12. doi:10.1016/j.hydromet.2015.07.008
Wang X, Chen Q, Yin Z, Zhang P, Long Z, Su Z (2003) Removal of impurities from copper electrolyte with adsorbent containing antimony. Hydrometallurgy 69:39–44. doi:10.1016/S0304-386X(03)00026-4
Ando K, Tsuchida N (1997) Recovering Bi and Sb from electrolyte in copper electrorefining. JOM 49:49–51. doi:10.1007/s11837-997-0033-4
Awe SA, Sundkvist JE, Bolin NJ, Sandstrom A (2013) Process flowsheet development for recovering antimony from Sb-bearing copper concentrates. Miner Eng 49:45–53. doi:10.1016/j.mineng.2013.04.026
Zhang B, Li Q, Shen W, Min X (2012) Recovery of bismuth and antimony metals from pressure-leaching slag. Rare Met 31:102–106. doi:10.1007/s12598-012-0471-1
Hoffmann J (2004) The purification of copper refinery electrolyte. JOM 56(7):30–33. doi:10.1007/s11837-004-0088-4
Möller CA, Myagmarsuren B, Friedrich B (2010) Effect of As, Sb, Bi and oxygen in copper anodes during electrorefining. In: Copper 2010: Proceedings, Hamburg, Germany
Deorkar NV, Tavlarides LL (1997) A chemically bonded adsorbent for separation of antimony, copper and lead. Hydrometallurgy 46:121–135. doi:10.1016/S0304-386X(97)00006-6
Navarro P, Alguacil FJ (2002) Adsorption of antimony and arsenic from a copper electrorefining solution onto activated carbon. Hydrometallurgy 66:101–105. doi:10.1016/S0304-386X(02)00108-1
Xiao F, Mao J, Cao D, Shen X, Volinsky AA (2012) The role of trivalent arsenic in removal of antimony and bismuth impurities from copper electrolytes. Hydrometallurgy 125–126:76–80. doi:10.1016/j.hydromet.2012.05.011
F-x Xiao, Cao D, J-w Mao, X-n Shen, F-z Ren (2013) Role of trivalent antimony in the removal of As, Sb, and Bi impurities from copper electrolytes. Int J Min Met Mater 20:9–16. doi:10.1007/s12613-013-0687-6
Szymanowski JAN (1998) Removal of toxic elements from copper electrolyte by solvent extraction. Miner Process Extr Metall 18:389–418. doi:10.1080/08827509808914162
Navarro P, Simpson J, Alguacil FJ (1999) Removal of antimony(III) from copper in sulphuric acid solutions by solvent extraction with LIX 1104SM. Hydrometallurgy 53:121–131. doi:10.1016/S0304-386X(99)00033-X
Sarkar SG, Dhadke PM (1999) Solvent extraction separation of antimony(III) and bismuth(III) with bis(2,4,4-trimethylpentyl) monothiophosphinic acid (Cyanex 302). Sep Purif Technol 15:131–138. doi:10.1016/S1383-5866(98)00088-4
Iyer JN, Dhadke PM (2003) Solvent extraction and separation studies of antimony(III) and bismuth(III) by using cyanex-925. Indian J Chem Technol 10:665–669
Fujii T, Aoki K, Yamana H (2007) Extraction behavior of antimony in a TRUEX system. J Nucl Sci Technol 44:1301–1305. doi:10.1080/18811248.2007.9711375
Lin HK (2004) Extraction of antimony by a copper chloride extractant. Hydrometallurgy 73:283–291. doi:10.1016/j.hydromet.2003.12.008
Dalton RF, Diaz G, Price R, Zunkel AD (1991) The cuprex metal extraction process: recovering copper from sulfide ores. JOM 43(8):51–56. doi:10.1007/BF03221105
Alian A, Sanad W (1967) Extraction of antimony with tertiary amines. Talanta 14:659–669. doi:10.1016/0039-9140(67)80033-X
Sargar BM, Rajmane MM, Anuse MA (2004) Selective liquid-liquid extraction of antimony(III) from hydrochloric acid media by N-n-octylaniline in xylene. J Serb Chem Soc 69:283–298
Facon S, Cote G, Bauer D (1991) Solvent extraction of antimony(III), bismuth (III), lead(II), tin(IV) with bis(2,4,4-trimethylpentyl)phosphinodithioic acid (Cyanex 301 ®). Solvent Extr Ion Exch 9:717–734. doi:10.1080/07366299108918080
Mineral Commodity Summaries (2015). U.S.A
Thornton I, Rautiu R, Brush M (2001) Chapter 4: lead industry profile. Lead the facts. Ian Allan Printing Ltd, Hersham, U.K., pp 47–65
Blanpain B, Arnout S, Chintinne M, Swinbourne DR (2014) Chapter 8—lead recycling. In: Worrell E, Reuter MA (eds) Handbook of recycling. Elsevier, Boston, MA, pp 95–111. doi:10.1016/B978-0-12-396459-5.00008-8
Liu W-F, Yang T-Z, Xia W-T, Xie Z-F, Liu W, Huang C (2009) Principle and practice of producing qualified antimony white from lead-antimony alloy by blowing directly. In: Proceedings of the symposium of the extraction and processing division, held at the TMS 2009 annual meeting and exhibition. TMS, Warrendale Pennsylvania, USA, pp. 483–492
Binz F, Friedrich B (2015) Recovery of antimony trioxide flame retardants from lead refining residues by slag conditioning and fuming. Chem Ing Tech 87:1569–1579. doi:10.1002/cite.201500071
Liu W, Yang T, Zhang D, Chen L, Liu Y (2014) A new pyrometallurgical process for producing antimony white from by-product of lead smelting. JOM 66(9):1694–1700. doi:10.1007/s11837-014-1026-8
Osmani A, Rizaj M, Terziqi A, Kamberaj N (2009) Slag Valorisation of reductive smelting process by shaft furnace in the lead metallurgy of “Trepça” complex with economical and environmental effects. J Int Environ Appl Sci 4:198–206
Singh LN (1990) Synthesis of potassium antimony tartrate from the antimony dross of lead smelters. Hydrometallurgy 25:19–25. doi:10.1016/0304-386X(90)90061-6
H-z Cao, J-z Chen, H-j Yuan, G-q Zheng (2010) Preparation of pure SbCl3 from lead anode slime bearing high antimony and low silver. Trans Nonferrous Met Soc China 20:2397–2403. doi:10.1016/S1003-6326(10)60661-9
Minić D, Štrbac N, Mihajlović I, Živković Ž (2005) Thermal analysis and kinetics of the copper-lead matte roasting process. J Therm Anal Calorim 82:383–388. doi:10.1007/s10973-005-0906-0
Barry DL, Cannell JF, Catterall JA, Chave PA, Harris M, Read R, Taylor MRG (1995) Metal manufacturing, refining and finishing works. Ruislip, UK
Jaeck ML (1989) Primary and secondary lead processing. In: Proceedings of the international symposium on primary and secondary lead processing. Halifax, Canada, Elsevier Science
Henry H (1926) Refining lead. U.S. Patent US1573830 A
Nikolić BG (1997) Processing of alkali antimony intermediate products in a lead refinery. Hydrometallurgy 47:31–36. doi:10.1016/S0304-386X(97)00023-6
Ojebuoboh FK (1996) One ton of bismuth in the lead smelter and 60 years of Kroll-Betteron. CIM Bull 89:76–79
Mallaley K, Morris DR (1990) Analysis of the Kroll-Betterton process—the removal of bismuth from lead bullion. Can Metal Q 29:67–71
Brecka G, Hein K, Lange HJ, Paschen P (1997) A pyrometallurgical alternative: the refining electrolysis of lead and solder. JOM 49(4):62–64. doi:10.1007/BF02914881
González-Domínguez JA, Peters E, Dreisinger DB (1991) The refining of lead by the Betts process. J Appl Electrochem 21:189–202. doi:10.1007/BF01052570
Felder A, Prengaman RD (2006) Lead alloys for permanent anodes in the nonferrous metals industry. JOM 58(10):28–31. doi:10.1007/s11837-006-0197-3
Peterson M, Twidwell LG (1985) Removal of arsenic from lead smelter speiss. J Hazard Mater 12:225–229. doi:10.1016/0304-3894(85)85009-3
Itoh S, Ono J, Hino M, Nagasaka T (2005) Kinetic study on recovery of antimony in anode slime from used lead batteries utilizing volatile oxide formation. Mater Trans 46:658–664. doi:10.2320/matertrans.46.658
Lin D, Qiu K (2011) Removal of arsenic and antimony from anode slime by vacuum dynamic flash reduction. Environ Sci Technol 45:3361–3366. doi:10.1021/es103424u
Qiu K, Lin D, Yang X (2012) Vacuum evaporation technology for treating antimony-rich anode slime. JOM 64:1321–1325. doi:10.1007/s11837-012-0458-2
Koparal AS, Özgür R, Öğütveren ÜB, Bergmann H (2004) Antimony removal from model acid solutions by electrodeposition. Sep Purif Technol 37:107–116. doi:10.1016/j.seppur.2003.09.001
Bergmann MEH, Koparal AS (2011) Electrochemical antimony removal from accumulator acid: results from removal trials in laboratory cells. J Hazard Mater 196:59–65. doi:10.1016/j.jhazmat.2011.08.073
Thiele UK (2004) Quo vadis polyester catalyst? Chem Fibers Int 54:162–163
Albonetti S, Blanchard G, Burattin P, Masetti S, Tagliani A, Trifirò F (1998) A new ternary mixed oxide catalyst for ammoxidation of propane: Sn/V/Sb. Catal Lett 50:17–23. doi:10.1023/A:1019046430749
Trimm DL, Gabbay DS (1971) Kinetics and mechanism of oxidation of butene isomers over a tin oxide + antimony oxide catalyst. T Faraday Soc 67:2782–2792. doi:10.1039/TF9716702782
Santacesaria E, Di Serio M, Basile G, Carra S (1989) Kinetics of chloroform fluorination by HF catalyzed by antimony pentachloride. J Fluorine Chem 44:87–111. doi:10.1016/S0022-1139(00)84373-0
Ono T, Hillig KW, Kuczkowski RL (1990) The mechanism of the oxidation of propene to acrolein over antimony-tin mixed oxide catalysts. J Catal 123:236–244. doi:10.1016/0021-9517(90)90172-G
Zhang C, Catlow CRA (2008) The mechanism of propene oxidation to acrolein on iron antimony oxide. J Catal 259:17–25. doi:10.1016/j.jcat.2008.06.027
Thiele UK (2001) The current status of catalysis and catalyst development for the industrial process of poly(ethylene terephthalate) polycondensation. Int J Polym Mater 50:387–394. doi:10.1080/00914030108035115
Biros SM, Bridgewater BM, Villeges-Estrada A, Tanski JM, Parkin G (2002) Antimony ethylene glycolate and catecholate compounds: structural characterization of polyesterification catalysts. Inorg Chem 41:4051–4057. doi:10.1021/ic020204b
Aharoni SM (1998) The cause of the grey discoloration of PET prepared by the use of antimony-catalysts. Polym Eng Sci 38:1039–1047. doi:10.1002/pen.10272
Takahashi Y, Sakuma K, Itai T, Zheng G, Mitsunobu S (2008) Speciation of antimony in PET bottles produced in Japan and China by X-ray absorption fine structure spectroscopy. Environ Sci Technol 42:9045–9050. doi:10.1021/es802073x
Fernschild G, Rudolph W, Massonne J (1977) Process for the recovery of antimony pentachloride from used catalyst solutions. U.S. Patent US4005176 A
Hyatt DE (1988) Recovery or arsenic and antimony from spent antimony catalyst. U.S. Patent US4722774 A
Van Brecht A, Konings A (2011) In: Jones PT et al (eds) Second international slag valorisation symposium: The transition to sustainable materials management. KU Leuven, Leuven, pp 215–227
Jung C-H, Osako M (2009) Metal resource potential of residues from municipal solid waste (MSW) melting plants. Resour Conserv Recy 53:301–308. doi:10.1016/j.resconrec.2009.01.004
Van Gerven T, Geysen D, Stoffels L, Jaspers M, Wauters G, Vandecasteele C (2005) Management of incinerator residues in Flanders (Belgium) and in neighbouring countries. A comparison. Waste Manage 25:75–87. doi:10.1016/j.wasman.2004.09.002
Nakamura K, Kinoshita S, Takatsuki H (1996) The origin and behavior of lead, cadmium and antimony in MSW incinerator. Waste Manage 16:509–517. doi:10.1016/S0956-053X(96)00093-1
Watanabe N, Inoue S, Ito H (1999) Antimony in municipal waste. Chemosphere 39:1689–1698. doi:10.1016/S0045-6535(99)00069-7
Osako M, Machida N, Tanaka M (1996) Risk management measures against antimony in residue after incineration of municipal waste. Waste Manage 16:519–526. doi:10.1016/S0956-053X(96)00103-1
Paoletti F, Seifert H, Vehlow J, Sirini P (2000) Oxyanions forming elements in waste combustion- partitioning of antimony. Waste Manage Res 18:141–150. doi:10.1177/0734242x0001800206
Miravet R, López-Sánchez JF, Rubio R (2006) Leachability and analytical speciation of antimony in coal fly ash. Anal Chim Acta 576:200–206. doi:10.1016/j.aca.2006.06.003
Van Gerven T, Cooreman H, Imbrechts K, Hindrix K, Vandecasteele C (2007) Extraction of heavy metals from municipal solid waste incinerator (MSWI) bottom ash with organic solutions. J Hazard Mater 140:376–381. doi:10.1016/j.jhazmat.2006.10.037
Okkenhaug G, Breedveld GD, Kirkeng T, Lægreid M, Mæhlum T, Mulder J (2013) Treatment of air pollution control residues with iron rich waste sulfuric acid: does it work for antimony (Sb)? J Hazard Mater 248–249:159–166. doi:10.1016/j.jhazmat.2012.12.041
Beard A, Klimes M (2013) Searching for safe flame retardants—an update on regulatory status and environmental assessments. PINFA and CEFIC, San Francisco, CA. http://www.flameretardants-online.com/images/userdata/pdf/375_EN.pdf
Camino G, Costa L (1988) Performance and mechanisms of fire retardants in polymers—a review. Polym Degrad Stab 20:271–294. doi:10.1016/0141-3910(88)90073-0
Camino G, Costa L, Luda di Cortemiglia MP (1991) Thermal stabilisation and flammability of polymers and composites: overview of fire retardant mechanisms. Polym Degrad Stab 33:131–154. doi:10.1016/0141-3910(91)90014-I
SRI-Consulting (2008) Global market of flame retardants. http://flameretardants-online.com/web/en/106/114.htm
Cheema HA, El-Shafei A, Hauser PJ (2013) Conferring flame retardancy on cotton using novel halogen-free flame retardant bifunctional monomers: synthesis, characterizations and applications. Carbohydr Polym 92:885–893. doi:10.1016/j.carbpol.2012.09.081
Abou-Okeil A, El-Sawy SM, Abdel-Mohdy FA (2013) Flame retardant cotton fabrics treated with organophosphorus polymer. Carbohydr Polym 92:2293–2298. doi:10.1016/j.carbpol.2012.12.008
Edwards B, El-Shafei A, Hauser P, Malshe P (2012) Towards flame retardant cotton fabrics by atmospheric pressure plasma-induced graft polymerization: synthesis and application of novel phosphoramidate monomers. Surf Coat Technol 209:73–79. doi:10.1016/j.surfcoat.2012.08.031
Costa L, Goberti P, Paganetto G, Camino G, Sgarzi P (1990) Thermal behaviour of chlorine-antimony fire-retardant systems. Polym Degrad Stab 30:13–28. doi:10.1016/0141-3910(90)90114-M
Keresztes S, Tatár E, Mihucz VG, Virág I, Majdik C, Záray G (2009) Leaching of antimony from polyethylene terephthalate (PET) bottles into mineral water. Sci Total Environ 407:4731–4735. doi:10.1016/j.scitotenv.2009.04.025
Belarra MA, Belategui I, Lavilla I, Anzano JM, Castillo JR (1998) Screening of antimony in PVC by solid sampling-graphite furnace atomic absorption spectrometry. Talanta 46:1265–1272. doi:10.1016/S0039-9140(97)00390-1
Minozaki M, Kachi M (1994) Method and apparatus for recovery of bromine and/or antimony from heat-resistant resins. Japan Patent JP 06144801 A
Masatochi J, Juji I, Daisuke A, Shinji S (1995) Treatment of decomposition gases of flame-resistant plastics. Japan Patent JP 08299759, A
Masatoshi I, Ikuta Y (1998) Pyrolysis-based material recovery from molding resin for electronic parts. J Environ Eng 124:821–828. doi:10.1061/(ASCE)0733-9372
Jakab E, Uddin MA, Bhaskar T, Sakata Y (2003) Thermal decomposition of flame-retarded high-impact polystyrene. J Anal Appl Pyrolysis 68–69:83–99. doi:10.1016/S0165-2370(03)00075-5
Klein J, Dorge S, Trouvé G, Venditti D, Durécu S (2009) Behaviour of antimony during thermal treatment of Sb-rich halogenated waste. J Hazard Mater 166:585–593. doi:10.1016/j.jhazmat.2008.12.006
Wu H, Shen Y, Harada N, An Q, Yoshikawa K (2014) Production of pyrolysis oil with low bromine and antimony contents from plastic material containing brominated flame retardants and antimony trioxide. Energy Environ Res 4:105–118. doi:10.5539/eer.v4n3p105
Mitan NMM, Bhaskar T, Hall WJ, Muto A, Williams PT, Sakata Y (2008) Effect of decabromodiphenyl ether and antimony trioxide on controlled pyrolysis of high-impact polystyrene mixed with polyolefins. Chemosphere 72:1073–1079. doi:10.1016/j.chemosphere.2008.04.011
Brebu M, Jakab E, Sakata Y (2007) Effect of flame retardants and Sb2O3 synergist on the thermal decomposition of high-impact polystyrene and on its debromination by ammonia treatment. J Anal Appl Pyrolysis 79:346–352. doi:10.1016/j.jaap.2007.02.003
Hall WJ, Mitan NMM, Bhaskar T, Muto A, Sakata Y, Williams PT (2007) The co-pyrolysis of flame retarded high impact polystyrene and polyolefins. J Anal Appl Pyrolysis 80:406–415. doi:10.1016/j.jaap.2007.05.002
Onwudili JA, Williams PT (2009) Alkaline reforming of brominated fire-retardant plastics: fate of bromine and antimony. Chemosphere 74:787–796. doi:10.1016/j.chemosphere.2008.10.029
Onwudili JA, Williams PT (2009) Degradation of brominated flame-retarded plastics (Br-ABS and Br-HIPS) in supercritical water. J Supercrit Fluids 49:356–368. doi:10.1016/j.supflu.2009.03.006
Lateef H, Grimes SM, Morton R, Mehta L (2008) Extraction of components of composite materials: ionic liquids in the extraction of flame retardants from plastics. J Chem Technol Biotechnol 83:541–545. doi:10.1002/jctb.1829
Freegard K, Tan G, Morton R (2006) Develop a process to separate brominated flame retardants from WEEE polymers: final reports. Oxon, UK
Wäger P, Böni H, Buser A, Morf L, Schluep M, Streicher M (2009) Recycling of plastics from waste electrical and electronic equipment (WEEE)—tentative results of a swiss study. In: R’09 Twin World Congress & World Resources Forum, Davos, Switzerland
Buekens A, Yang J (2014) Recycling of WEEE plastics: a review. J Mater Cycles Waste 16:415–434. doi:10.1007/s10163-014-0241-2
Guo J, Guo J, Xu Z (2009) Recycling of non-metallic fractions from waste printed circuit boards: a review. J Hazard Mater 168:567–590. doi:10.1016/j.jhazmat.2009.02.104
de Marco I, Caballero BM, Chomón MJ, Laresgoiti MF, Torres A, Fernández G, Arnaiz S (2008) Pyrolysis of electrical and electronic wastes. J Anal Appl Pyrolysis 82:179–183. doi:10.1016/j.jaap.2008.03.011
Moltó J, Font R, Gálvez A, Conesa JA (2009) Pyrolysis and combustion of electronic wastes. J Anal Appl Pyrolysis 84:68–78. doi:10.1016/j.jaap.2008.10.023
Gullett BK, Linak WP, Touati A, Wasson SJ, Gatica S, King CJ (2007) Characterization of air emissions and residual ash from open burning of electronic wastes during simulated rudimentary recycling operations. J Mater Cycles Waste 9:69–79. doi:10.1007/s10163-006-0161-x
Guo Q, Yue X, Wang M, Liu Y (2010) Pyrolysis of scrap printed circuit board plastic particles in a fluidized bed. Powder Technol 198:422–428. doi:10.1016/j.powtec.2009.12.011
Morf LS, Tremp J, Gloor R, Huber Y, Stengele M, Zennegg M (2005) Brominated flame retardants in waste electrical and electronic equipment: substance flows in a recycling plant. Environ Sci Technol 39:8691–8699. doi:10.1021/es051170k
Hall WJ, Williams PT (2006) Fast pyrolysis of halogenated plastics recovered from waste computers. Energy Fuels 20:1536–1549. doi:10.1021/ef060088n
Hall WJ, Miskolczi N, Onwudili J, Williams PT (2008) Thermal processing of toxic flame-retarded polymers using a waste fluidized catalytic cracker (FCC) catalyst. Energy Fuels 22:1691–1697. doi:10.1021/ef800043g
Tostar S, Stenvall E, Boldizar A, Foreman MRSJ (2013) Antimony leaching in plastics from waste electrical and electronic equipment (WEEE) with various acids and gamma irradiation. Waste Manage 33:1478–1482. doi:10.1016/j.wasman.2013.03.002
Zhenming X, Hongzhou L, Jia L, Yongqing W (2006) Vacuum distillation method for recovering antimony from waste printed circuit board. China Patent CN 1760385, A
Yang X, Sun L, Xiang J, Hu S, Su S (2013) Pyrolysis and dehalogenation of plastics from waste electrical and electronic equipment (WEEE): a review. Waste Manage 33:462–473. doi:10.1016/j.wasman.2012.07.025
Belardi G, Ippolito N, Piga L, Serracino M (2014) Investigation on the status of rare earth elements contained in the powder of spent fluorescent lamps. Thermochim Acta 591:22–30. doi:10.1016/j.tca.2014.07.015
Chang T-C, Wang S-F, You S-J, Cheng A (2007) Characterization of halophosphate phosphor powders recovered from the spent fluorescent lamps. J Environ Eng Manage 17:435–439
Henderson ST, Marsden AM, Thorn Lighting L (1972) Lamps and lighting: a manual of lamps and lighting. Edward Arnold, London
Shinde K, Dhoble SJ, Swart HC, Park K (2012) Some halophosphates phosphors. Phosphate phosphors for solid-state lighting, vol 174. Springer, Berlin, pp 151–189. doi:10.1007/978-3-642-34312-4_6
Srivastava AM, Sommerer TJ (1998) Fluorescent lamp phosphors. Electrochem Soc Interface 7:28–31
Shionoya S, Yen WM, Yamamoto H (2006) Phosphor handbook. Taylor & Francis, Boca Raton, FL
Soules TF (2000) Tailoring lamp phosphors. In: Ronda CR, Shea LE, Srivastava AM (eds) Physics and chemistry of luminescent materials, vol 40. Electrochemical Society, Pennington, NJ, pp 56–68
Braconnier JJ, Rollat A (2012) Method for recovering rare-earth elements from a solid mixture containing a halophosphate and a compound of one or more rare-earth elements. European Patent EP 2419377 A1
Dobrowolski R, Mierzwa J (1993) Investigation of activator (Mn, Sb) speciation in phosphors for fluorescent lamps. Mater Chem Phys 34:270–273. doi:10.1016/0254-0584(93)90046-O
Baliga BJ (2015) The IGBT device: physics, design and applications of the insulated gate bipolar transistor. Elsevier Science - William Andrew, Norwich, NY
Compact fluorescent lamps could nearly halve global lighting demand for electricity (2013). Worldwatch Institute, Washington, DC. http://www.worldwatch.org/node/5918
Binnemans K, Jones PT (2014) Perspectives for the recovery of rare earths from end-of-life fluorescent lamps. J Rare Earth 32:195–200. doi:10.1016/S1002-0721(14)60051-X
Binnemans K, Jones PT, Blanpain B, Van Gerven T, Yang Y, Walton A, Buchert M (2013) Recycling of rare earths: a critical review. J Clean Prod 51:1–22. doi:10.1016/j.jclepro.2012.12.037
Innocenzi V, De Michelis I, Kopacek B, Vegliò F (2014) Yttrium recovery from primary and secondary sources: a review of main hydrometallurgical processes. Waste Manage 34:1237–1250. doi:10.1016/j.wasman.2014.02.010
Liu H, Zhang S, Pan D, Tian J, Yang M, Wu M, Volinsky AA (2014) Rare earth elements recycling from waste phosphor by dual hydrochloric acid dissolution. J Hazard Mater 272:96–101. doi:10.1016/j.jhazmat.2014.02.043
Mei G, Rao P, Mitsuaki M, Toyohisa F (2009) Separation of red (Y2O3: Eu3+), blue (Sr, Ca, Ba)10(PO4)6Cl2: Eu2+ and green (LaPO4: Tb3+, Ce3+) rare earth phosphors by liquid/liquid extraction. J Wuhan Univ Technol 24:418–423. doi:10.1007/s11595-009-3418-0
Tunsu C, Ekberg C, Retegan T (2014) Characterization and leaching of real fluorescent lamp waste for the recovery of rare earth metals and mercury. Hydrometallurgy 144–145:91–98. doi:10.1016/j.hydromet.2014.01.019
Urniezaite I, Denafas G, Jankunaite D (2009) Characterization of residues from physicochemical treatment of waste fluorescent lamps. Waste Manage Res 28:606–614. doi:10.1177/0734242x09341074
Wu Y, Yin X, Zhang Q, Wang W, Mu X (2014) The recycling of rare earths from waste tricolor phosphors in fluorescent lamps: a review of processes and technologies. Resour Conserv Recy 88:21–31. doi:10.1016/j.resconrec.2014.04.007
Yang F, Kubota F, Baba Y, Kamiya N, Goto M (2013) Selective extraction and recovery of rare earth metals from phosphor powders in waste fluorescent lamps using an ionic liquid system. J Hazard Mater 254–255:79–88. doi:10.1016/j.jhazmat.2013.03.026
Yang HL, Wang W, Cui HM, Zhang DL, Liu Y, Chen J (2012) Recovery of rare earth elements from simulated fluorescent powder using bifunctional ionic liquid extractants (Bif-ILEs). J Chem Technol Biotechnol 87:198–205. doi:10.1002/Jctb.2696
Dupont D, Binnemans K (2015) Rare-earth recycling using a functionalized ionic liquid for the selective dissolution and revalorization of Y2O3:Eu3+ from lamp phosphor waste. Green Chem 17:856–868. doi:10.1039/C4GC02107J
Dupont D, Binnemans K (2016) Antimony recovery from the halophosphate fraction in lamp phosphor waste: a zero-waste approach. Green Chem 18:175–185. doi:10.1039/C5GC01746G
Tian H et al (2014) A comprehensive global inventory of atmospheric antimony emissions from anthropogenic activities, 1995–2010. Environ Sci Technol 48:10235–10241. doi:10.1021/es405817u
Wilson SC, Lockwood PV, Ashley PM, Tighe M (2010) The chemistry and behaviour of antimony in the soil environment with comparisons to arsenic: a critical review. Environ Pollut 158:1169–1181. doi:10.1016/j.envpol.2009.10.045
Varrica D, Bardelli F, Dongarrà G, Tamburo E (2013) Speciation of Sb in airborne particulate matter, vehicle brake linings, and brake pad wear residues. Atmos Environ 64:18–24. doi:10.1016/j.atmosenv.2012.08.067
von Uexküll O, Skerfving S, Doyle R, Braungart M (2005) Antimony in brake pads—a carcinogenic component? J Clean Prod 13:19–31. doi:10.1016/j.jclepro.2003.10.008
Filella M, Belzile N, Chen Y-W (2002) Antimony in the environment: a review focused on natural waters: I. Occurrence. Earth-Sci Rev 57:125–176. doi:10.1016/S0012-8252(01)00070-8
Ungureanu G, Santos S, Boaventura R, Botelho C (2015) Arsenic and antimony in water and wastewater: overview of removal techniques with special reference to latest advances in adsorption. J Environ Manage 151:326–342. doi:10.1016/j.jenvman.2014.12.051
Kang M, Kamei T, Magara Y (2003) Comparing polyaluminum chloride and ferric chloride for antimony removal. Water Res 37:4171–4179. doi:10.1016/S0043-1354(03)00351-8
Wu Z, He M, Guo X, Zhou R (2010) Removal of antimony (III) and antimony (V) from drinking water by ferric chloride coagulation: competing ion effect and the mechanism analysis. Sep Purif Technol 76:184–190. doi:10.1016/j.seppur.2010.10.006
Guo X, Wu Z, He M (2009) Removal of antimony(V) and antimony(III) from drinking water by coagulation–flocculation–sedimentation (CFS). Water Res 43:4327–4335. doi:10.1016/j.watres.2009.06.033
Xu W, Wang H, Liu R, Zhao X, Qu J (2011) The mechanism of antimony(III) removal and its reactions on the surfaces of Fe–Mn Binary Oxide. J Colloid Interface Sci 363:320–326. doi:10.1016/j.jcis.2011.07.026
Biswas BK, J-i Inoue, Kawakita H, Ohto K, Inoue K (2009) Effective removal and recovery of antimony using metal-loaded saponified orange waste. J Hazard Mater 172:721–728. doi:10.1016/j.jhazmat.2009.07.055
Wang L, C-l Wan, Zhang Y, Lee D-J, Liu X, X-f Chen, Tay J-H (2015) Mechanism of enhanced Sb(V) removal from aqueous solution using chemically modified aerobic granules. J Hazard Mater 284:43–49. doi:10.1016/j.jhazmat.2014.10.041
Dou X, Mohan D, Zhao X, Pittman CU Jr (2015) Antimonate removal from water using hierarchical macro-/mesoporous amorphous alumina. Chem Eng J 264:617–624. doi:10.1016/j.cej.2014.11.123
Dong S, Dou X, Mohan D, Pittman CU Jr, Luo J (2015) Synthesis of graphene oxide/schwertmannite nanocomposites and their application in Sb(V) adsorption from water. Chem Eng J 270:205–214. doi:10.1016/j.cej.2015.01.071
Zhu J, Wu F, Pan X, Guo J, Wen D (2011) Removal of antimony from antimony mine flotation wastewater by electrocoagulation with aluminum electrodes. J Environ Sci 23:1066–1071. doi:10.1016/S1001-0742(10)60550-5
Acknowledgments
The authors wish to thank the KU Leuven (Projects GOA/13/008 and IOF-KP RARE3) and the FWO Flanders (PhD fellowship to DD) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
The contributing editor for this article was Diran Apelian.
Rights and permissions
About this article

Cite this article
Dupont, D., Arnout, S., Jones, P.T. et al. Antimony Recovery from End-of-Life Products and Industrial Process Residues: A Critical Review. J. Sustain. Metall. 2, 79–103 (2016). https://doi.org/10.1007/s40831-016-0043-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-016-0043-y