
Abstract
Introduction
Psoriatic arthritis (PsA) is an immune-mediated chronic inflammatory arthropathy associated with impaired physical function and reduced quality of life. Biologic therapies that target tumor necrosis factor (anti-TNF) have significantly improved clinical outcomes. Partial, non- and transient responses remain common comprising significant unmet clinical need. New therapies with novel modes of action are urgently required.
Objectives
The interleukin (IL)-17 pathway has recently been attributed a critical role in the pathogenesis of spondyloarthritides. Herein, we review data from clinical studies with secukinumab, a novel fully human IgG1κ anti-IL-17A monoclonal antibody (mAb), in patients with active PsA.
Results
Across two pivotal phase 3 studies, secukinumab provided significant and sustained reductions in the signs and symptoms of PsA, inhibition of radiographic progression, and improved patient-reported outcomes and measures of quality of life. The primary efficacy endpoint, a ≥20% improvement from baseline according to the American College of Rheumatology 20 (ACR20) response at Week 24, was significantly higher in patients treated with secukinumab compared with placebo, with improvements sustained through at least 52 weeks. Clinical benefits were seen with secukinumab regardless of concomitant methotrexate treatment and in patients who were either anti-TNF-naïve or who were inadequate responders to anti-TNF therapy. Secukinumab was well-tolerated, with a safety profile consistent with that previously reported in psoriasis trials. The most common adverse events were nasopharyngitis, upper respiratory tract infections, and headache.
Conclusion
Secukinumab offers an effective new addition to the available treatment options for PsA. Regulatory submissions have been filed worldwide, with the first approvals recently obtained in Japan and Europe. Future studies are required to define the optimal timing and strategic use of this novel treatment modality.
Funding
Novartis Pharma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Psoriatic arthritis (PsA) is an immune-mediated chronic inflammatory arthropathy-associated with psoriasis [1]. The occurrence of PsA has been estimated at between 6% and 42% of patients afflicted with psoriasis [2–6], which occurs in 1–3% of the population [7–9]. The clinical manifestations of PsA include arthritis, enthesitis, dactylitis, spondylitis, psoriasis, and nail disease, with approximately 50% of patients also experiencing erosive joint damage within the first 2 years [10]. Thus, therapeutic principles should comprise management of disease in each tissue compartment, with the ultimate goal of impeding disease progression and maximizing function over time [11, 12].
Until the advent of tumor necrosis factor (TNF) inhibitors, treatment for PsA focused largely on the use of non-steroidal anti-inflammatory drugs (NSAIDs) and conventional disease-modifying anti-rheumatic drugs (DMARDs) despite a dearth of evidence showing their efficacy in clinical trials [13]. Nevertheless, clinical experience suggests that these agents are effective at reducing inflammation and treating some symptoms and signs and they remain the recommended first-line treatment option for patients with active PsA across several current international guidelines [11, 12].
The development of anti-TNF therapies revolutionized the treatment of PsA, offering an effective biologic treatment for patients who showed a lack of efficacy and/or toxicity with NSAIDs and DMARDs [11, 14–16]. Extensive evidence accrued with several agents across numerous clinical trials and registries shows that anti-TNF agents are efficacious in the treatment of PsA. However, a number of unmet needs remain; for example, some patients have an inadequate response to, or intolerance of anti-TNF agents, long-term therapy with these agents is associated with decreasing drug survival rates, and the increased risk of infection may be of concern to some patients [16–18].
Consequently, therapies with differing modes of action, including agents that were developed for the treatment of other rheumatic diseases (e.g., abatacept, rituximab, tocilizumab), have been tested for potential efficacy in PsA [16, 19]. Some of these agents, such as the phosphodiesterase 4 (PDE4) inhibitor apremilast [20] and the interleukin (IL)-12/23 inhibitor ustekinumab [21], have demonstrated efficacy in clinical trials and are approved for use in PsA in the majority of developed countries. Nevertheless, because of inadequate efficacy, intolerance, or safety issues new treatments with alternative modes of action continue to be sought. Recently, genome-wide association studies, together with translational immunology analyses, have identified several novel molecular cascades involved in the pathogenesis of psoriasis and PsA, including particularly the IL-17 pathway [22].
This article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
The IL-17 Pathway in PsA
The inflammatory cytokine IL-17 is part of a family of six homodimeric cytokines (IL-17A–F) and one heterodimer (IL-17A/F), that in turn signal via five IL-17 receptors (IL-17RA–E, in turn functioning as heterodimers). Discrete IL-17 family members elaborate functional and immunological differences [23–26]. IL-17A is a dimeric glycoprotein that functions in both the innate and adaptive immune responses, and specifically mediates effects in antibacterial and fungal immunity and tissue repair [23, 27]. The effects of the other members of the IL-17 family are less well characterized, although evidence suggests they are involved in antimicrobial defense through the innate immune response. IL-17A is produced by a range of immune cells, including especially Th17 cells and Type 3 innate lymphoid cells and can affect the function of several cell types such as neutrophils, keratinocytes, fibroblast-like synoviocytes, endothelial cells, chondrocytes, and osteoblasts [28–30]. Activation of these cells results in the release of further pro-inflammatory cytokines and chemokines, which aside from promoting inflammation, drive other pathological processes including hyperproliferation, matrix destruction, vessel activation, bone erosion, and cartilage damage [28–30].
The IL-17 pathway has been shown to play an important role in the pathology of PsA [18, 31]. Inflammation in PsA is characterized by synovial tissue enriched by expression of IL-17A and IL-17RA [32]. Whereas CD4+ T cells express IL-17A, IL-17A-producing CD8+ T cells are more abundant in the synovial fluid of patients with PsA, compared to healthy individuals, and the levels of these cells positively correlate with measures of disease activity and joint damage progression [33]. Evidence from preclinical studies further supports the contribution of the IL-17 pathway to PsA pathogenesis. Local overexpression of IL-17 during collagen-induced arthritis in mice is associated with increased synovial inflammation and joint destruction [34], while elevated expression of IL-23 in a murine model induced a population of IL-23R+CD3+CD4−CD8− entheseal resident T cells to produce inflammatory mediators including IL-17 and IL-22, which was accompanied by histologic evidence of enthesitis [34, 35].
Consequently, biological therapies targeting the IL-17 pathway, including antibodies against IL-17A (secukinumab and ixekizumab) and IL-17RA (brodalumab), have been extensively evaluated in psoriasis, PsA, and other spondyloarthritides [29].
Secukinumab: Mode of Action, Pharmacokinetics, and Pharmacodynamics
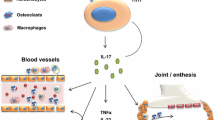
Secukinumab is a fully human IgG1κ anti-IL-17A monoclonal antibody (mAb), which targets the function of IL-17A [36]. Secukinumab selectively binds to and neutralizes IL-17A, inhibiting its interaction with IL-17 receptors expressed on keratinocytes, fibroblast-like synoviocytes, endothelial cells, chondrocytes, and osteoblasts. As a result, secukinumab inhibits downstream inflammatory pathways implicated in autoimmune and inflammatory diseases, while leaving other immune functions undisturbed (Fig. 1) [29, 36–39].

Secukinumab prevents IL-17A binding to its receptor, inhibiting production of pro-inflammatory mediators [54]. IFN interferon, IL interleukin
In patients with psoriasis, serum levels of total IL-17A (free and secukinumab-bound IL-17A) increase to plateau serum concentrations after administration of secukinumab. Following cessation of treatment, serum levels slowly decrease reflecting the kinetics of clearance of secukinumab-bound IL-17A. No significant changes in IL-17F are seen after secukinumab treatment, indicating that secukinumab selectively binds to and neutralizes free IL-17A [40]. In patients with plaque psoriasis, infiltrating epidermal neutrophils and various neutrophil-associated markers were significantly reduced in lesional skin of plaque psoriasis patients after 1–2 weeks of treatment with secukinumab, compared with baseline [40].
The pharmacokinetic (PK) profile of secukinumab in patients with PsA is typical for an IgG1 mAb and similar to that seen in patients with psoriasis [40]. Based on population PK analysis in patients with plaque psoriasis, after initial weekly dosing during the first month, time to reach the maximum concentration of secukinumab was 31–34 days. Based on simulated data, peak concentrations at steady state (Cmax ss) following subcutaneous (sc) administration of secukinumab 300 or 150 mg were estimated at 55.2 and 27.6 µg/mL, respectively, after 20 weeks. Secukinumab is absorbed with an average absolute bioavailability of 73%, with a volume of distribution of 7.10–8.60 L following a single intravenous (i.v.) dose, suggesting that limited distribution to peripheral compartments occurs. Mean systemic clearance is 0.19 L/day, and is not influenced by gender, dose, or time. The mean elimination half-life of secukinumab in patients with psoriasis is 27 days. In patients with PsA, the bioavailability of secukinumab is 85%, and although clearance and volume of distribution increase as body weight increases, clearance is independent of age.
Although no interaction studies have been performed in humans, there is no evidence to indicate that IL-17A will influence the expression of CYP450 enzymes [40]. The formation of some CYP450 enzymes is suppressed by increased levels of cytokines during chronic inflammation. Thus, conceivably, agents targeting the IL-17 pathway may result in ‘normalization’ of CYP450 levels with accompanying lower exposure of CYP450-metabolized concomitant medications. No interaction was seen when secukinumab was administered together with methotrexate (MTX) and/or corticosteroids in PsA studies [40].
Clinical Trials of Secukinumab in PsA
In a small phase II proof-of-concept study in patients with PsA, secukinumab showed improvements in clinical response, C-reactive protein (CRP) level, erythrocyte sedimentation rate (ESR), and quality of life (QoL) measures versus placebo, although it should be noted that the primary endpoint, a ≥20% improvement from baseline according to the American College of Rheumatology 20 (ACR20) response at Week 6, was not met [41]. Based on these promising preliminary findings, two large, randomized, placebo-controlled phase III studies involving more than 1000 patients with active PsA, FUTURE 1 (ClinicalTrials.gov identifier: NCT01392326; [42]) and FUTURE 2 (ClinicalTrials.gov identifier: NCT01752634; [43]), were subsequently initiated.
FUTURE 1 and FUTURE 2
In FUTURE 1, patients with active PsA were randomized (1:1:1) to one of three treatment arms: i.v. secukinumab 10 mg/kg (weeks 0, 2, 4) followed by sc secukinumab 150 or 75 mg every 4 weeks, or placebo (Fig. 2a). In FUTURE 2, patients were randomized (1:1:1:1) to one of four arms: sc secukinumab 300, 150, 75 mg, or placebo once a week from baseline to Week 4 and then every 4 weeks thereafter (Fig. 2b). In both studies, placebo patients switched to sc secukinumab 150 or 75 mg at Week 16 or 24, depending upon their clinical response. In both FUTURE 1 and FUTURE 2, patients who had an inadequate response to TNF inhibitors (anti-TNF-IR), or who had not previously received TNF inhibitors (anti-TNF-naïve), were eligible. At baseline, approximately two-thirds of patients were anti-TNF-naïve and around half were receiving concomitant MTX (Table 1). Both studies are ongoing with FUTURE 1 planned to run for 2 years followed by a 3-year extension study and FUTURE 2 for the initial 52 weeks of study followed by an additional 4 years during which long-term efficacy and safety data will be collected.

Study designs of FUTURE 1 (a) and FUTURE 2 (b)
The primary endpoint in both FUTURE 1 and FUTURE 2 was ACR20 response at Week 24 [42, 43]. This time point was chosen to align with the assessment of radiographic progression [modified total Sharp score (mTSS)] at Week 24 in the FUTURE 1 study and for consistency across both studies. Other secondary endpoints were Psoriasis Area Severity Index (PASI)75 and PASI90, Disease Activity Score (DAS)28-CRP, SF-36 Physical Component Summary (PCS) score, Health Assessment Questionnaire Disability Index (HAQ-DI), ACR50 response, and presence of dactylitis and enthesitis. Pre-specified exploratory endpoints included ACR70 responses, additional patient-reported outcomes and subgroup analyses according to previous anti-TNF use.
Key Results
Across FUTURE 1 and FUTURE 2, secukinumab provided rapid and clinically meaningful improvements in multiple facets of PsA, including joint symptoms, skin symptoms, dactylitis, and enthesitis. The primary and all pre-defined secondary endpoints were met with both secukinumab doses in FUTURE 1 (Figs. 3, 4, 5, 6, 7, 8, 9, 10) [42]. At Week 24, the ACR20 response rates were 50.0% with secukinumab i.v.-150 mg, 50.5% with secukinumab i.v.-75 mg, and 17.3% with placebo (P < 0.001 for both comparisons versus placebo; Fig. 3). ACR50/70 responses at Week 24 were 34.7%/18.8%, 30.7%/16.8%, and 7.4%/2.0%, respectively (all P < 0.001; Fig. 4). The clinical improvements offered by secukinumab in FUTURE 1 were sustained with long-term therapy. Applying a conservative estimate of efficacy with missing values imputed as non-response, ACR20 responses at Week 52 were 59.9% and 56.9%, respectively [42]. Observed values of ACR20 responses at Week 52 were 69.5% with secukinumab 150 mg and 66.9% with secukinumab 75 mg (Fig. 3). Two-year follow-up data from FUTURE 1 confirmed the sustainability of these effects with long-term secukinumab treatment [42].

Figure 3a from The New England Journal of Medicine. Mease PJ, McInnes IB, Kirkham B, et al. 2015; 373:1329-1339. Copyright © 2015 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society
ACR20 responses from baseline to Week 24 (placebo-controlled period), and through Week 52 (observed) for patients randomized to secukinumab at baseline. a Proportion of patients achieving ACR20 in FUTURE 1. b Proportion of patients achieving ACR20 in FUTURE 2. *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Missing values were imputed as nonresponse (nonresponder imputation) up to Week 24. Observed data from Week 28–52

Figure 4a from The New England Journal of Medicine. Mease PJ, McInnes IB, Kirkham B, et al. 2015; 373:1329-1339. Copyright © 2015 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society
Proportion of patients achieving ACR50 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Missing values were imputed as nonresponse (nonresponder imputation) up to Week 24. Observed data from Week 28–52

Proportion of patients achieving PASI75 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Missing values were imputed as nonresponse (nonresponder imputation) up to Week 24. Observed data from Week 28–52

Proportion of patients achieving PASI90 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Missing values were imputed as nonresponse (nonresponder imputation) up to Week 24. Observed data from Week 28–52

Mean change from baseline in DAS-28 through Week 52 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Means are from mixed-effect model repeated measures shown up to Week 24. Observed data from Week 28–52

Mean change from baseline in SF-36 PCS through Week 52 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Means are from mixed-effect model repeated measures shown up to Week 24. Observed data from Week 28–52

Mean change from baseline in HAQ-DI through Week 52 in FUTURE 1 (a) and FUTURE 2 (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing). Means are from mixed-effect model repeated measures shown up to Week 24. Observed data from Week 28–52

Mean changes in total mTSS, erosion, and JSN scores from baseline to Week 24 in FUTURE 1 (full analysis set). ‡ P < 0.05 vs. placebo (P values at Week 24 adjusted for multiplicity of testing); Linear extrapolation of missing values was applied at Week 24
In FUTURE 2, the primary endpoint was met with all secukinumab doses [43]. ACR20 response rates at Week 24 were 54.0% with secukinumab 300 mg (P < 0.0001), 51.0% with 150 mg (P < 0.0001), and 29.3% with 75 mg (P = 0.0399) versus 15.3% with placebo (Fig. 3). Mean changes from baseline in DAS28-CRP and SF36-PCS were all significantly improved with secukinumab 300 and 150 mg versus placebo at Week 24 (Figs. 7, 8). Secukinumab 300 mg also significantly improved ACR50 and HAQ-DI versus placebo (Figs. 4, 9). Other endpoints were not considered significant based on hierarchical testing. ACR50 responses were achieved by 35.0%, 35.0%, and 18.2% of patients in the secukinumab 300, 150, and 75 mg groups, respectively, compared with 7.1% in the placebo group (Fig. 4). The clinical improvements induced by secukinumab in FUTURE 2 were sustained through to Week 52; response rates were 64.0%, 64.0%, and 50.5% with missing values imputed as nonresponse in the secukinumab 300, 150, and 75 mg groups, respectively. Observed ACR20 responses were 73%, 73%, and 67%, respectively (Fig. 3) [43].
Psoriasis
Consistent with its clearly demonstrated effect in primary cutaneous psoriasis phase III clinical trials, secukinumab has been shown to significantly improve the symptoms and signs of both skin and nail psoriasis in patients with PsA. Significant improvements were seen in PASI75/90 responses at Week 24 in both FUTURE 1 and FUTURE 2 versus placebo (Figs. 5, 6), with responses sustained for up to 52 weeks [42, 43]. Furthermore, improvements in Investigator’s Global Assessment (IGA) 0/1 and mean Nail Psoriasis Severity Index (NAPSI) scores, reductions in high-sensitivity C-reactive protein (hsCRP) levels, and clinically meaningful improvements (≥4-point change from baseline) in Dermatology Life Quality Index (DLQI) were also seen with secukinumab versus placebo at Week 24 in both studies [44, 45].
Radiographic Progression
Secukinumab significantly inhibited radiographic progression of PsA versus placebo at Week 24 in FUTURE 1, as assessed by the mTSS (Fig. 10), with this effect sustained through 52 weeks of treatment [46]. Improvements in mTSS were reflected by improvements in both the erosion and joint-space-narrowing scores (Fig. 10). Among those patients who had initially received placebo for the first 24 weeks then switched to secukinumab, radiographic progression was also inhibited. At Week 24, the proportion of patients with no disease progression was greater for the secukinumab groups compared with placebo-treated patients (82.3% and 92.3% for the i.v.-150 mg and i.v.-75 mg, respectively, vs. 75.7% for the placebo group). This effect was sustained in the secukinumab groups at Week 52 (85.7% and 85.8%), and increased in patients who were initially randomized to placebo and then given active treatment (86.8%).
Dactylitis and Enthesitis
In FUTURE 1, a significantly greater proportion of patients achieved complete resolution of dactylitis and enthesitis with secukinumab (pooled doses; predefined secondary endpoint) compared with placebo at Week 24. Exploratory analysis of dactylitis and enthesitis resolution in the individual dose arms of FUTURE 1 was consistent with the pooled analysis. In patients with symptoms at baseline, 48.1% and 46.0% in the i.v.-150 mg group and 56.7% and 48.8% in the i.v.-75 mg group showed complete resolution of dactylitis and enthesitis, respectively, at Week 24, versus 15.5% and 12.8% with placebo (all P < 0.0001). At Week 52, 87.7% and 89.7% of patients in the i.v.-150 mg and i.v.-75 mg groups, respectively, were free from dactylitis compared with 48.5% and 48.5% at baseline (Fig. 11); 81.6% and 79.4% were free from enthesitis at Week 52 versus 37.6% and 36.1% at baseline.

Resolution of enthesitis and dactylitis at Week 24 in those patients with symptoms at baseline in FUTURE 1 and FUTURE 2 (a). The proportions of patients without enthesitis and dactylitis at baseline and Week 52 are also shown (b). *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo. Missing values were imputed as nonresponse (nonresponder imputation) at Weeks 24; observed data are shown at Week 52. (P values at Week 24 adjusted for multiplicity)
Although numerical improvements in the proportion of patients achieving resolution of dactylitis and enthesitis with pooled secukinumab versus placebo were seen in FUTURE 2, these changes were not statistically significant when examined by a hierarchical testing methodology. Nevertheless, exploratory analysis of dactylitis and enthesitis resolution by individual doses indicated clinically meaningful improvements in resolution with secukinumab 300 mg and 150 mg versus placebo at Week 24 [47]. In patients with symptoms at baseline, 56.5% (P < 0.01) and 48.2% (P < 0.01) in the 300 mg group and 50.0% (P < 0.01) and 42.2% (P < 0.05) in the 150 mg group showed complete resolution of dactylitis and enthesitis, respectively, at Week 24, versus 14.8% and 21.5% with placebo. At Week 52, 88.2% and 90.9% of patients in the 300 and 150 mg groups, respectively, were free from dactylitis compared with 54.0% and 68.0% at baseline (Fig. 11); 72.0% and 69.3% were free from enthesitis at Week 52 versus 44.0% and 36.0% at baseline. Secukinumab also reduced the number of dactylitic digits and enthesitis sites, determined via multiple methods of assessment at Week 24.
Patient-Reported Outcomes
Secukinumab also mediated a positive impact on various patient-reported outcomes including physical function, QoL and fatigue. In FUTURE 1, SF-36 PCS and HAQ-DI were significantly improved with both secukinumab i.v.-150 mg and i.v.-75 mg versus placebo at Week 24, while in FUTURE 2, SF-36 PCS was significantly enhanced with secukinumab 300 and 150 mg and HAQ-DI was significantly improved with secukinumab 300 mg (Figs. 8, 9). Exploratory analyses of several other patient-reported outcomes in FUTURE 2 at Week 24 also showed significant improvements in fatigue, as measured by the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scale, and QoL, as measured by the Psoriatic Arthritis Quality of Life (PsA QoL) score, for secukinumab versus placebo [43, 48].
Efficacy of Secukinumab in Patient Subgroups
Pre-specified subgroup analyses of FUTURE 1 and FUTURE 2 showed clinical benefits with secukinumab in patients who were anti-TNF treatment-naïve and patients who were prior inadequate responders to anti-TNF (TNFi-exposed). In both studies, significantly greater proportions of patients achieved ACR responses with secukinumab versus placebo after 24 weeks in both subgroups [42, 43]. In FUTURE 1, ACR20 response rates in the TNF-naïve subgroup using nonimputed data (missing values were imputed as non-response [non-responder imputation (NRI)] at Week-24 were 54.5% for secukinumab i.v.-150 mg and 55.6% for secukinumab i.v.-75 mg, compared with 17.5% in the placebo group. At Week 52 using observed data, the ACR20 response rates were 75.2% for secukinumab i.v.-150 mg and 73.2% for secukinumab i.v.-75 mg (Fig. 12a). In the TNFi-exposed subgroup, the ACR20 responses at Week 24 were 39.0% and 38.3% in the secukinumab i.v.-150 mg and i.v.-75 mg groups, respectively, compared with 16.9% in the placebo group. The Week 52 observed data were 53.3% for secukinumab i.v.-150 mg and 51.0% for secukinumab i.v.-75 mg (Fig. 12a).

ACR20 and ACR50 response with secukinumab by anti-TNF status in FUTURE 1 (a) and FUTURE 2 (b) at Week 24 and Week 52. Missing values were imputed as non-response (non-responder imputation) at Week 24; observed data are shown at Week 52. *P < 0.0001; † P < 0.001; ‡ P < 0.05 vs. placebo
Similarly, in FUTURE 2, ACR20 response rates in the TNF-naïve subgroup using nonimputed data at Week 24 were 58.2% for secukinumab 300 mg, 63.5% for secukinumab 150 mg, 36.9% for secukinumab 75 mg, and 15.9% in the placebo group. At Week 52 using observed data, the ACR20 response rates were 79.4% for secukinumab 300 mg, 84.7% for secukinumab 150 mg, and 67.9% for secukinumab 75 mg (Fig. 12b). In the TNFi-exposed subgroup, the ACR20 responses at Week 24 were 45.5%, 29.7% and 14.7% in the secukinumab 300, 150, and 75 mg groups, respectively, compared with 14.3% in the placebo group. The Week 52 observed data were 62.1% for secukinumab 300 mg, 37.8% for secukinumab 150 mg, and 63.2% for secukinumab 75 mg (Fig. 12b).
Improvements with secukinumab versus placebo were also seen in both subgroups in a range of other endpoints, including PASI75/90, DAS-28, and HAQ-DI [42, 43, 49]. The magnitude of response was generally higher in the anti-TNF-naïve population [42, 43]. A dose effect emerged in this context, analogous to that observed in cutaneous psoriasis studies, since secukinumab 300 mg was associated with the greatest improvements in anti-TNF-IR patients. The efficacy of secukinumab was sustained for up to 52 weeks in both subgroups [49]. Finally, it is worth noting that in post hoc analyses, improvements in ACR response rates with secukinumab compared with placebo at Week 24 were similar regardless of concomitant MTX use [42, 43].
Safety in Phase III Trials
Secukinumab was well-tolerated in patients with active PsA across FUTURE 1 and FUTURE 2 (Table 2) [42, 43]. More than 1200 patients were assessed for safety across the two FUTURE trials, with a mean exposure to secukinumab of 438.5 days in FUTURE 1 and 411.7 days in FUTURE 2 (Table 2). Among these patients, the exposure-adjusted incidence rate of any adverse event in the secukinumab-treated patients for FUTURE 1 and FUTURE 2 was 471 and 307 per 100 patient-years, respectively (Table 2). Across both studies, the most common adverse events were nasopharyngitis, upper respiratory tract infections, and headache (Table 2).
Serious adverse events were also uncommon among secukinumab-treated patients. Discontinuation due to adverse events occurred in 23 (3.9%) secukinumab-treated patients in FUTURE 1 and 8 (2.1%) secukinumab-treated patients in FUTURE 2, compared with 5 (2.5%) and 4 (4.1%) patients in the placebo groups of FUTURE 1 and FUTURE 2, respectively.
Adverse events of special interest were similar between groups in both studies (Table 2). Three patients (0.7%) had Candida infection in the FUTURE 1 study compared with no patients in the placebo group in the first 16 weeks. In FUTURE 2, 2 patients (0.7%) had Candida infection compared with no patients in the placebo group. There was only one death, due to intracranial venous sinus thrombosis in FUTURE 1. Adverse events of inflammatory bowel disease/Crohn’s disease were rare with secukinumab. One patient in the placebo group and one patient in the secukinumab group experienced de novo events of Crohn’s disease across the entire treatment period of FUTURE 1. There were 12 cases of neutropenia in secukinumab-treated patients across the entire treatment period of FUTURE 1 and 5 cases in secukinumab-treated patients in FUTURE 2; the majority of events were transient Grade 1 or 2 neutropenia and there were no instances of Grade 4 neutropenia. No patients withdrew from either FUTURE 1 or FUTURE 2 because of neutropenia. Incidences of major adverse cardiac events and malignancy were also low among secukinumab-treated patients. No attempted or completed suicide or suicidal ideation was reported in secukinumab-treated patients across both studies.
Secukinumab has a low potential for immunogenicity, as evidenced by the low incidence of treatment-emergent anti-drug antibodies (ADAs; i.e., tests that were positive during the study but negative at baseline). In FUTURE 1, while 3 out of 10 patients with ADAs at baseline continued to have ADAs and neutralizing antibodies in all or most post-baseline samples, treatment-emergent ADAs were detected in only one patient. Similarly, in FUTURE 2 treatment-emergent ADAs were detected in one patient who switched to secukinumab 150 mg at week 24, but no immunogenicity-related adverse events or loss of efficacy were reported in this patient.
Secukinumab in The PsA Treatment Paradigm
Although head-to-head trials would be required to reach definite conclusions, indirect comparisons suggest that secukinumab is at least as effective as currently available therapies mediated via an alternative mode of action. Thus, secukinumab should be a useful addition to the PsA treatment armamentarium. Indeed, recent updates to treatment guidelines for PsA presented by the European League Against Rheumatism (EULAR) and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) have incorporated anti-IL-17 therapy into their treatment algorithms [11, 12]. Biologic therapy, including anti-TNF agents (e.g., infliximab, etanercept, certolizumab, golimumab and adalimumab), anti-IL-12/23 (e.g., ustekinumab) and anti-IL-17 therapy (e.g., secukinumab) is recommended for patients with inadequate response or intolerance to NSAIDs and conventional synthetic DMARDs. Biologics have demonstrated efficacy across PsA clinical domains, including peripheral arthritis, enthesitis, dactylitis, axial disease, or severe skin disease occurring in combination, particularly if the latter is dominant [11, 12].
Secukinumab (Cosentyx®, Novartis) was recently approved in Europe (October 2015) and in the United States (January 2016) for the treatment of active PsA. The secukinumab clinical trial program is ongoing, including three further phase 3 studies in PsA: FUTURE 3 (ClinicalTrials.gov identifier: NCT01989468) is a 24-week study that will investigate the safety and efficacy of subcutaneous secukinumab 300 and 150 mg versus placebo; FUTURE 4 (ClinicalTrials.gov identifier: NCT02294227) is a 16-week study that will assess the efficacy and safety of subcutaneous secukinumab 150 mg versus placebo with or without a loading regimen; and FUTURE 5 (ClinicalTrials.gov identifier: NCT02404350) will investigate the effect of subcutaneous secukinumab 150 and 300 mg on progression of structural damage for up to 2 years.
Currently, secukinumab is also the only IL-17A inhibitor approved as a first-line systemic treatment for moderate to severe plaque psoriasis in adult patients in Europe, and as a treatment for moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy in the US. Secukinumab has also been approved in Switzerland, Chile, Australia, Argentina, Canada, Japan, and Singapore for the treatment of moderate to severe plaque psoriasis. Approval was granted based on the significant efficacy shown with secukinumab in improving the signs and symptoms of psoriasis compared with placebo, etanercept, and ustekinumab in phase III trials [50, 51].
Secukinumab is being investigated across other rheumatic diseases. In patients with active ankylosing spondylitis (AS), secukinumab rapidly and significantly improved the signs and symptoms of disease versus placebo in two phase III trials [52] and was recently approved for the treatment of AS by the EMA and FDA. To our knowledge it is not being developed as a mono-biologic for patients with rheumatoid arthritis.
Conclusions
Secukinumab is the first anti-IL-17A therapy to demonstrate efficacy in phase 3 clinical trials in PsA. In more than 1000 patients across FUTURE 1 and FUTURE 2, significant efficacy was demonstrated with secukinumab versus placebo in all components of PsA including joint symptoms, skin symptoms, dactylitis, enthesitis, and patient-reported outcomes, with responses sustained up to 52 weeks. Furthermore, no radiographic disease progression was observed in more than 80% of the patients receiving secukinumab. Efficacy was demonstrated regardless of concomitant MTX therapy and in both anti-TNF-IR patients and those naïve to anti-TNF therapies. The safety profile of secukinumab was consistent with previous studies in psoriasis, with no new or unexpected safety findings identified.
These results highlight the important role played by IL-17A in the pathogenesis of PsA, and together with the positive results from recent studies in AS [53], suggest that secukinumab will be a valuable addition to the available treatment options for PsA and other chronic and disabling rheumatic diseases.
Change history
25 July 2016
An erratum to this article has been published.
References
Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64 Suppl 2:ii14–7.
Mease PJ, Gladman DD, Papp KA, Khraishi MM, Thaci D, Behrens F, Northington R, Fuiman J, Bananis E, Boggs R, Alvarez D. Prevalence of rheumatologist-diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. J Am Acad Dermatol. 2013;69:729–35.
Biondi Oriente C, Scarpa R, Pucino A, Oriente P. Psoriasis and psoriatic arthritis. Dermatological and rheumatological co-operative clinical report. Acta Derm Venereol Suppl (Stockh). 1989;146:69–71.
Scarpa R, Oriente P, Pucino A, Torella M, Vignone L, Riccio A, Biondi Oriente C. Psoriatic arthritis in psoriatic patients. Br J Rheumatol. 1984;23:246–50.
Shbeeb M, Uramoto KM, Gibson LE, O’Fallon WM, Gabriel SE. The epidemiology of psoriatic arthritis in Olmsted County, Minnesota, USA, 1982–1991. J Rheumatol. 2000;27:1247–50.
Stern RS. The epidemiology of joint complaints in patients with psoriasis. J Rheumatol. 1985;12:315–20.
Schafer T. Epidemiology of psoriasis. Review and the German perspective. Dermatology (Basel, Switzerland). 2006;212:327–37.
Raychaudhuri SP, Farber EM. The prevalence of psoriasis in the world. J Euro Acad Dermatol Venereol JEADV. 2001;15:16–7.
Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512–6.
Kane D, Stafford L, Bresnihan B, FitzGerald O. A prospective, clinical and radiological study of early psoriatic arthritis: an early synovitis clinic experience. Rheumatology (Oxford, England). 2003;42:1460–8.
Gossec LSJ, Ramiro S, de Wit M, Cutolo M, Dougados M, Emery P, Landewé R, Oliver S, Aletaha D, Betteridge N, Braun J, Burmester G, Cañete JD, Damjanov N, FitzGerald O, Haglund E, Helliwell P, Kvien TK, Lories R, Luger T, Maccarone M, Marzo-Ortega H, McGonagle D, McInnes IB, Olivieri I, Pavelka K, Schett G, Sieper J, van den Bosch F, Veale DJ, Wollenhaupt J, Zink A, van der Heijde D. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75:499–510.
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta Felquer M, Armstrong AW, Bautista Molano W, Boehncke WH, Campbell W, Cauli A, Espinoza LR, FitzGerald O, Gladman DD, Gottlieb A, Helliwell PS, Elaine Husni M, Love TJ, Lubrano E, McHugh N, Nash P, Ogdie A, Orbai AM, Parkinson A, O’Sullivan D, Rosen CF, Schwartzman S, Siegel EL, Toloza S, Tuong W, Ritchlin CT. Group for research and assessment of psoriasis and psoriatic arthritis: treatment recommendations for psoriatic arthritis 2015. Arthritis Rheumatol. 2016. doi:10.1002/art.39573 (Epub ahead of print).
Li Y, Wang D, Wang Y, Shi G. Progress of biological agents on psoriatic arthritis. Curr Pharm Biotechnol. 2014;15:525–34.
Boehncke WH, Menter A. Burden of disease: psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2013;14:377–88.
Eder L, Thavaneswaran A, Chandran V, Gladman DD. Tumour necrosis factor alpha blockers are more effective than methotrexate in the inhibition of radiographic joint damage progression among patients with psoriatic arthritis. Ann Rheum Dis. 2014;73:1007–11.
Mease PJ. Biologic therapy for psoriatic arthritis. Rheum Dis Clin North Am. 2015;41:723–38.
Lebwohl MG, Bachelez H, Barker J, Girolomoni G, Kavanaugh A, Langley RG, Paul CF, Puig L, Reich K, van de Kerkhof PC. Patient perspectives in the management of psoriasis: results from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis Survey. J Am Acad Dermatol. 2014;70:871–881.e1-30.
Marinoni B, Ceribelli A, Massarotti MS, Selmi C. The Th17 axis in psoriatic disease: pathogenetic and therapeutic implications. Auto-immunity Highl. 2014;5:9–19.
Mease PJ, Armstrong AW. Effective management of psoriasis and psoriatic arthritis: insights on current and emerging therapies and enhanced professional collaboration. Semin Arthritis Rheum. 2014;44:e7–8.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, Lespessailles E, Hall S, Hochfeld M, Hu C, Hough D, Stevens RM, Schett G. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73:1020–6.
Kavanaugh A, Ritchlin C, Rahman P, Puig L, Gottlieb AB, Li S, Wang Y, Noonan L, Brodmerkel C, Song M, Mendelsohn AM, McInnes IB. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis. 2014;73:1000–6.
Bluett J, Barton A. What have genome-wide studies told us about psoriatic arthritis? Curr Rheumatol Rep. 2012;14:364–8.
Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67.
Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–74.
Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, Tomkinson KN, Fitz LJ, Wolfman NM, Collins M, Dunussi-Joannopoulos K, Chatterjee-Kishore M, Carreno BM. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol. 2008;181:2799–805.
Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17:435–40.
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–76.
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–98.
Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery. 2012;11:763–76.
Smith JA, Colbert RA. Review: the interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: Th17 and beyond. Arthritis Rheumatol (Hoboken, NJ). 2014;66:231–41.
Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008;58:2307–17.
Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359:419–29.
Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, Taams LS, Kirkham BW. Interleukin-17+ CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol (Hoboken, NJ). 2014;66:1272–81.
Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van den Berg WB. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–13.
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, Eberl G, Buckley CD, Kastelein RA, Pierce RH, Laface DM, Cua DJ. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+ CD4−CD8− entheseal resident T cells. Nat Med. 2012;18:1069–76.
Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72 Suppl 2:ii116–23.
Sanford M, McKeage K. Secukinumab: first global approval. Drugs. 2015;75:329–38.
Gaffen SL, Kramer JM, Yu JJ, Shen F. The IL-17 cytokine family. Vitam Horm. 2006;74:255–82.
Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, Russell CB. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol. 2013;133:17–26.
Novartis. Cosentyx summary of product characteristics. http://ec.europa.eu/health/documents/community-register/2015/20150115130444/anx_130444_en.pdf. Accessed 7 Dec 2015.
McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, Dahmen G, Wollenhaupt J, Schulze-Koops H, Kogan J, Ma S, Schumacher MM, Bertolino AP, Hueber W, Tak PP. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73:349–56.
Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewe R, Nash P, Pricop L, Yuan J, Richards HB, Mpofu S. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. 2015;373:1329–39.
McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, van der Heijde D, Landewe R, Conaghan PG, Gottlieb AB, Richards H, Pricop L, Ligozio G, Patekar M, Mpofu S. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;386:1137–46.
Gottlieb AB, Mease P, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, Nash P, Pricop L, Yuan J, Richards H, Mpofu S. Secukinumab, a human anti-interleukin-17A monoclonal antibody, significantly reduces psoriasis burden in patients with psoriatic arthritis: results from a phase 3 randomized controlled trial. Arthritis Rheumatol. 2014;66:S233.
Gottlieb A, McInnes IB, Mease P, Rahman P, Kandala S, Patekar M, Mpofu S. THU0418 secukinumab significantly reduces psoriasis burden in patients with psoriatic arthritis: results from the phase 3 future 2 study. Ann Rheum Dis. 2015;74:349–50.
van der Heijde D, Landewe R, Mease P, McInnes I, Conaghan P, Pricop L, Ligozio G, Richards H, Mpofu S. Secukinumab inhibits radiographic progression in patients with psoriatic arthritis: data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study (FUTURE 1). Ann Rheum Dis. 2015;74(Suppl 2):347.
Kirkham B, McInnes IB, Mease P, Kremer J, Kandala S, Pricop L, Mpofu S. THU0421 secukinumab is effective in reducing dactylitis and enthesitis using multiple measures in patients with psoriatic arthritis: data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study (future 2). Ann Rheum Dis. 2015;74:351.
Rahman P, Strand V, McInnes I, Marzo-Ortega H, Dokoupilova E, Churchill M, Kandala S, Pricop L, Mpofu S. Secukinumab improves physical function, quality of life, fatigue and work productivity in patients with active psoriatic arthritis in FUTURE 2, a Phase 3 trial. Ann Rheum Dis. 2015;74:356.
Kavanaugh A, McInnes I, Hall S, Chinoy H, Kivitz A, Kandala S, Patekar M, Mpofu S. Secukinumab efficacy in anti-TNF-naive and anti-TNF-IR patients with psoriatic arthritis: results of a phase 3 multicenter, double-blind, placebo-controlled study (FUTURE 2). Ann Rheum Dis. 2015;74:345.
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326–38.
Thaci D, Blauvelt A, Reich K, Tsai TF, Vanaclocha F, Kingo K, Ziv M, Pinter A, Hugot S, You R, Milutinovic M. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400–9.
Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, Deodhar A, Porter B, Martin R, Andersson M, Mpofu S, Richards HB. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373:2534–48.
Duarte JH. Spondyloarthropathies: IL-17A blockade ameliorates ankylosing spondylitis. Nat Rev Rheumatol. 2016;12:72.
Ivanov S, Linden A. Interleukin-17 as a drug target in human disease. Trends Pharmacol Sci. 2009;30:95–103.
Acknowledgments
Iain B. McInnes acknowledges support from Arthritis Research UK for the ongoing rheumatology program in University of Glasgow. Medical writing and editorial support in the development of this manuscript was provided by Tina Patrick, PhD, of Novartis Global Medical & Clinical Services, Dublin, Ireland and Ben Drever of Ashfield Healthcare Ltd. Support for this assistance was funded by Novartis Pharma. Philip Mease and Iain B. McInnes meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. The article processing charges for this publication were funded by Novartis Pharma.
Disclosures
Iain B. McInnes has received honoraria and research funding from Novartis, Eli-Lilly, Pfizer, Abbvie, MSD, and Janssen. Philip Mease has received research funding, consultation and/or speaking honoraria from Abbvie, Amgen, BMS, Boehringer Ingelheim, Celgene, Crescendo Bioscience, Genentech, Janssen, Lilly, Merck, Novartis, Pfizer, and UCB.
Compliance with Ethics Guidelines
This article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/06B4F0604C95F427.
An erratum to this article is available at https://doi.org/10.1007/s40744-016-0039-x.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mease, P., McInnes, I.B. Secukinumab: A New Treatment Option for Psoriatic Arthritis. Rheumatol Ther 3, 5–29 (2016). https://doi.org/10.1007/s40744-016-0031-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-016-0031-5