
Abstract
Purpose of Review
Several environmental contaminants have been implicated as contributors to COVID-19 susceptibility and severity. Immunomodulation and epigenetic regulation have been hypothesized as mediators of this relationship, but the precise underlying molecular mechanisms are not well-characterized. This review examines the evidence for epigenetic modification at the intersection of COVID-19 and environmental chemical exposures.
Recent Findings
Numerous environmental contaminants including air pollutants, toxic metal(loid)s, per- and polyfluorinated substances, and endocrine disrupting chemicals are hypothesized to increase susceptibility to the SARS-CoV-2 virus and the risk of severe COVID-19, but few studies currently exist. Drawing on evidence that many environmental chemicals alter the epigenetic regulation of key immunity genes and pathways, we discuss how exposures likely perturb host antiviral responses. Specific mechanisms vary by contaminant but include general immunomodulation as well as regulation of viral entry and recognition, inflammation, and immunologic memory pathways, among others.
Summary
Associations between environmental contaminants and COVID-19 are likely mediated, in part, by epigenetic regulation of key immune pathways involved in the host response to SARS-CoV-2.
Similar content being viewed by others

Introduction
The novel coronavirus SARS-CoV-2 has infected hundreds of millions of individuals worldwide, causing coronavirus disease 2019 (COVID-19) to become a global pandemic. In early 2020, epidemiologic research aimed at identifying COVID-19 risk factors began to occur at a rapid pace. Since that time, non-pharmaceutical interventions and vaccines have been recognized as key to slowing transmission [1, 2]. However, despite these measures, new cases and deaths have continued to accrue. While still evolving, the current understanding of contributors to COVID-19 severity includes aging, obesity, hypertension, chronic inflammation, and other clinical factors [3,4,5]. Additionally, there is increasing evidence suggesting that environmental chemicals play a role. For example, data from England show air pollution exposures may be a risk factor, with nitrogen oxide (NOX) and particulate matter (PM) levels associated with 3–12% more COVID-19 cases, and NOx levels associated with 3% more deaths [6••]. Precisely how environmental chemical exposures affect human susceptibility to COVID-19 and the course of infection remains uncertain, but undoubtedly involve numerous immune pathways.
The fact that environmental factors can influence the immune system is well-established. The discipline of immunotoxicology dates to the early 1970s [7] and has repeatedly demonstrated that many environmental chemicals stimulate and/or suppress innate, humoral, and/or cell-mediated immune responses [8]. Yet, the clinical sequelae of such immunotoxicity are complex and considerable gaps remain in the knowledge of interactions between pathogens, immune responses, and environmental exposures. Prior to the current pandemic, a workshop held by the National Academies of Sciences, Engineering, and Medicine emphasized the need for transdisciplinary and translational research to address such gaps, especially molecular mechanisms [9]. Disentangling the molecular pathways by which environmental chemical exposures increase susceptibility to or exacerbate infectious diseases is paramount to identifying causal factors and facilitating interventions that prevent or reduce morbidity and mortality.
The aim of this review is to examine the evidence linking environmental chemical exposures with viral infections (including COVID-19) and to highlight immune system pathways that may be under epigenetic control as potential intermediates. Of note, the studies compiled here do not encompass indirect pathways through which exposures to environmental chemicals increase the risk for chronic diseases that predispose individuals to severe COVID-19, as the evidence that certain environmental chemicals are endocrine disruptors [10] and/or carcinogenic [11] is beyond the scope of the current review.
Summary of Immune Pathways Involved in COVID-19 Infection, Progression, and Vaccination
SARS-CoV-2 can lead to dysregulation of the innate (non-specific) and adaptive (specific) arms of the immune systems, subverting natural defense mechanisms and resulting in severe disease [12••, 13••]. A complex inflammatory response is a hallmark of COVID-19 hospitalization and death [14,15,16]. Although immunopathology is not the focus of this article, a basic summary of the relevant immune pathways involved in COVID-19 is critical to understand how environmental chemical exposures and epigenetic perturbations may contribute.
SARS-CoV-2 Entry to the Cell
The angiotensin converting enzyme 2 (ACE2) is a part of the renin-angiotensin system and is a major entry point for the SARS-CoV-2 virus [17, 18, 19••, 20]. ACE2 is highly expressed in the lung and binds to the spike protein on the SARS-CoV-2 virus, creating a biological vulnerability to infection [17, 18, 19••]. Higher levels of ACE2 expression are correlated with greater susceptibility to SARS-CoV-2 infection and viral loads [15]. Following attachment to the ACE2 receptor, the host protein transmembrane serine protease 2 (TMPRSS2) activates the spike protein of SARS-CoV-2, facilitating membrane fusion that allows the virus to spread in lung tissue [20]. As such, greater TMPRSS2 expression has also been associated with severe COVID-19 [21].
Innate and Adaptive Immune Responses Following Infection
Following infection, pattern recognition receptors (PRRs), including toll-like receptors (TLRs), play a critical role in the early innate immune response [12••, 22]. TLRs recognize pathogens and trigger the release of interferon (IFN) proteins that “interfere” with viral replication [12••]. This pathway is initially delayed and diminished by SARS-CoV-2, which is unusually effective at reducing IFN production [13••]. The initial innate immune response via IFNs can profoundly impact the course of the disease: a stronger response can limit viral replication and quickly activate the adaptive immune system, whereas a weaker response can allow for rapid viral replication, delayed recruitment of the adaptive immune system, and hyperinflammation [13••, 22]. When adequate signaling occurs after infection to activate the adaptive immune system, B cells, CD4 + T cells, and CD8 + T cells are recruited to aid in controlling disease progression [13••]. Early recruitment typically results in a mild or asymptomatic case that does not necessitate hospitalization [13••]. Variation in the strength of both the innate and adaptive immune responses, therefore, contributes to the disparate outcomes observed following infection [13••].
Cytokine Alterations and Inflammatory Cell Death
Severe COVID-19 often involves a “cytokine storm,” an overproduction of pro-inflammatory proteins that leads to excessive systemic inflammation [12••]. The flood of pro-inflammatory cytokines primarily includes IFNs, interleukins (ILs), and tumor necrosis factor alpha (TNF-α) [12••, 23]. It is proposed that the underlying cause of cytokine overproduction is a weak or delayed adaptive immune response, which forces the body to “catch up” in fighting the infection [13••]. SARS-CoV-2 initiates the cytokine storm via the combination of tumor necrosis factor (TNF)-α and IFN-γ, resulting in the coordination of three distinct cell death pathways: pyroptosis, apoptosis, and necroptosis, together called PANoptosis [12••, 24]. PANoptosis induces further cytokine production, exacerbating the hyperinflammatory response and, in some cases, initiating a runaway cycle that can cause respiratory failure, exhaustion, and death [12••]. These findings highlight the complexity of the inflammatory response to SARS-CoV-2, demonstrating the vast dysregulation possible [15].
Changes in Immune Cell Composition
In addition to producing systemic inflammation, COVID-19 is associated with altered immune cell composition. The innate and the adaptive arms of the immune system are composed of distinct leukocyte subtypes. Cells of the innate immune system include granulocytes, monocytes, macrophages, and natural killer cells, among others, whereas cells of the adaptive immune system include B and T lymphocytes [25]. In severe cases of COVID-19, increased neutrophil and decreased lymphocyte counts are often observed, suggesting disruption of the immune cell milieu as an additional contributor to critical symptoms and death [14,15,16, 26,27,28].
Immunologic Memory
Following infection or vaccination, the adaptive immune system drives the induction of immunologic memory via B and T lymphocytes [13••]. The B cells secrete immunoglobulin M (IgM), G (IgG), and A (IgA) antibodies that neutralize the virus or viral infected cells [29], while the T cells support antibody production in addition to killing infected cells [30]. B and T cell responses and the longevity of resulting antibodies and immune memory can vary significantly between individuals [13••].
Epigenetic Perturbations of Immune Pathways by Environmental Contaminant Exposures
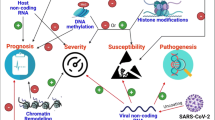
Genetics are one potential driver of susceptibility to SARS-CoV-2 infection. In a 2021 genome-wide association meta-analysis consisting of nearly 50,000 participants from across the globe, 13 loci were identified as being associated with SARS-CoV-2 infection, critical illness, or hospitalization due to COVID-19 [31]. By contrast, two recent epigenome-wide association studies (EWASs) revealed methylation levels at over 13,000 5’-C-phosphate-G-3’ (CpG) sites that were associated with infection status and 44 CpG sites that were associated with hospitalization requiring respiratory support [32••, 33••, 34], suggesting that epigenetic mechanisms also contribute to the immune responses to SARS-CoV-2. Epigenetic mechanisms modify gene expression without changing the underlying DNA sequence [34] and, in addition to DNA methylation at CpG sites, include modifications to histones (proteins in the cell nucleus that DNA is wrapped around) and non-coding microRNA (miRNA) [34]. Although each of these mechanisms is critical to regulating gene expression, histone modifications are difficult to characterize at the population-level and therefore are not frequently studied, whereas measuring DNA methylation and miRNA is feasible and increasingly being performed to better understand systems biology. Herein, we will focus on research involving DNA methylation and miRNA as potential intermediates linking classes of environmental chemicals with COVID-19 susceptibility and severity (Fig. 1).

Visual framework for how associations between environmental contaminants and COVID-19 may be mediated by epigenetic regulation of immune pathways, with a specific focus on DNA methylation and microRNAs. Abbreviations: PFAS = per- and polyfluorinated substances; EDCs = endocrine disrupting chemicals; miRNAs = microRNAs
Air Pollution
Air pollution is one of the most well-studied environmental contaminants in relation to COVID-19, partially because of wide data availability and well-known effects on the immune and respiratory systems. Air pollution is a general term that can include fine particulate matter—often categorized by diameter size as less than 2.5 µM (PM2.5) or less than 10 µM (PM10)—as well as nitrogen oxide (NOX), ozone (O3), polycyclic aromatic hydrocarbons (PAHs), volatile organic compounds (VOCs), sulfur oxides (SOX), and other contaminants. Epidemiological studies have established broad links between air pollutants and COVID-19 infections and deaths, particularly for PM and NOX [6••, 35,36,37,38,39]. Mechanistically, air pollution may alter COVID-19 susceptibility partially through epigenetic modification of immune system pathways.
As described above, it has been established that the SARS-CoV-2 virus utilizes the ACE2 receptor and TMPRSS2 to enter and infect cells, with greater expression of ACE2 and TMPRSS2 associated with increased infection and viral load [15, 19••, 20, 21]. ACE2 and TMPRSS2 expressions have been shown to increase following air pollutant exposure, specifically PM [19••, 40]. Additionally, ACE2 and TMPRSS2 are genes known to be regulated by DNA methylation, histone modification, and miRNAs, indicating that an individual’s epigenome may relate to ACE2 and TMPRSS2 expression [19••, 41, 42]. Recently, a study investigating methylation levels of ACE2 in nasal cells found differences between sex, race/ethnicity, and biological aging, with study authors indicating that nasal exposures to airborne environmental contaminants may be partially responsible for the observed changes [43•]. Nasal cells are exposed to air pollutants known to alter methylation signatures, some of which may be relevant to COVID-19 susceptibility [43•, 44]. While no known study has performed a targeted analysis on ACE2 and TMPRSS2 epigenetic changes following air pollution, EWASs and tangential evidence indicate that this connection may be possible [43•, 44].
Air pollutants are also known to alter many of the immune pathways involved in COVID-19 development after infection, partially explaining the associations observed between exposure and COVID-19 severity. A few specific pathways and key genes are worthy of further discussion: inflammation, TLRs, IFNs, and forkhead box P3 (FOXP3). Exposure to air pollutants is associated with the generation of reactive oxygen species (ROS) and pulmonary inflammation, resulting in adverse health effects such as asthma and decreased lung function [45, 46]. Chronic exposure to air pollutants results in degradation of multiple body systems, including the respiratory and immune systems that are largely involved in COVID-19 response [47, 48]. This inflammatory response is partially mediated by TLRs, a key portion of the physiological response to SARS-CoV-2 [12••, 13••, 49]. Exposure to PM has been associated with altered methylation levels of TLR genes, potentially altering the expression of these important immune receptors [50, 51]. Disruption of some TLRs predispose individuals to severe COVID-19, highlighting the need for further investigation of epigenetic regulation as a potential mechanism. In addition to inflammation and TLRs, air pollutants interact with IFNs, which are also known to play a large role in recruiting an effective immune response to COVID-19. Exposure to air pollution has been shown to increase methylation of IFN-γ, a gene involved in combatting viral replication and triggering inflammation [52, 53]. Although IFN-γ (type II) is not as robustly associated with COVID-19 development as type I and III IFNs, these findings nevertheless present a possibility for changes relevant to COVID-19 risk [12••]. Finally, ambient air pollution is known impair the function of regulatory T cells via hypermethylation of the FOXP3 gene. Regulatory T cells function to suppress immune response, and impairment of this process may make hyperinflammation more likely, a hallmark of severe COVID-19 [54].
Lastly, emerging evidence shows that greater air pollutant exposure is associated with lower titers of neutralizing antibodies to the SARS-CoV-2 virus following vaccination [55]. Similar associations were observed with other vaccines, and it is suggested that air-pollution-induced chronic inflammation may mediate associations with vaccine efficacy [55, 56]. We propose that epigenetics may play a role in this, regulating the increased inflammatory and decreased antibody responses.
Toxic Metal(loid)s
Arsenic (As), cadmium (Cd), mercury (Hg), and lead (Pb) are considered among the most significant potential threats to human health due to their toxicity and ubiquity [57]. As a result of both natural and anthropogenic processes, toxic metals and metalloids are widespread throughout the environment and are present in the air, water, soil, and food supply. For most individuals, exposures occur via ingestion of contaminated foods and drinking water, but exposure can also stem from cigarette smoke, industrial emissions, and certain occupations [58].
Lead is among the most well-studied toxic metal with respect to immunological effects following chronic exposures. Numerous epidemiologic studies have reported associations with increased susceptibility to infections [59, 60] and inflammation [61, 62], as well as altered humoral [63] and altered cell-mediated immunity [64,65,66,67]. Recently, there has been evidence to suggest that greater exposures to lead increase the risk for severe COVID-19, with severe cases excreting more lead in their urine relative to mild-to-moderate cases [68•]. One potential mechanism underlying lead-associated COVID-19 severity is differential methylation of runt-related (RUNX) transcription factor genes. In a 2015 study of children aged 3 months to 5 years from Detroit, blood lead levels were found to correlate with differential methylation of RUNX1 and RUNX3 [69]. These genes are essential for hematopoiesis (the process of blood cell formation), especially the development of B and T cells involved in the adaptive immune response [70]. Divergent B and T cell responses to SARS-CoV-2 have been hypothesized to be a key contributor to infection severity, as patients requiring intensive care unit (ICU) admission often exhibit an impaired CD4 + T cell response coupled with elevated antibody titers as compared to patients with more mild disease [71]. COVID-19 patients requiring ICU care are also more likely to be male [72], raising questions about sex-based differences in molecular pathways linking environmental exposures, epigenetics, and immune responses to SARS-CoV-2. Interestingly, the effects of lead exposure on methylation of RUNX genes may be sex-specific, with an inverse association observed for RUNX1 in males and a positive association observed for RUNX3 in females [69]. Future studies examining the role of toxic metal exposures in relation to COVID-19 severity should consider biologic sex as a potential modifying factor.
Other research points to the metalloid inorganic arsenic as a potent immunotoxicant with relevance for COVID-19. While studies have not yet investigated how arsenic is specifically related to SARS-CoV-2 infections, studies have linked higher exposures with other respiratory infections [73, 74, 75••]. Exposures that occur while in utero appear to be particularly important. In humans, prenatal arsenic exposures are associated with an increased risk of developing respiratory infections during the first year of life [73, 74]. In animals, the effects of prenatal arsenic exposures persist into adulthood, as evidenced by a 2020 study of mice exposed to arsenic in utero and later infected with the influenza A virus as adults [75••]. Compared to control mice, the arsenic exposed mice had greater lung damage and inflammation [75••]. Interestingly, the investigators noted that the exposed mice were not more likely to become infected with the influenza A virus [75••], and therefore reason that earlier epidemiologic studies may have been capturing respiratory infections that were more symptomatic [73, 74]. They propose that prenatal arsenic exposures influence the course of infectious disease progression, rather than susceptibility to infection per se. In support of this premise, the 2020 in vivo study additionally demonstrated that the hyperinflammatory response to the influenza A virus manifested with dysregulation of innate immune function of monocyte-derived macrophages, neutrophils, natural killer cells, and alveolar macrophages [75••]. This distinct phenotype is consistent with observational research showing epigenetic effects of prenatal arsenic exposure in human populations. As a specific example, profiling of cord blood from newborns prenatally exposed to arsenic via maternal ingestion of contaminated drinking water in Gómez Palacio, Mexico, revealed increased expression of 12 miRNAs with known roles in inflammatory response [76]. Of the 12 altered miRNAs, nine were predicted to target 66 mRNAs involved in TLR, nuclear factor kappa B (NF-kB), and IFN signaling pathways [76]. Within the context of COVID-19, alterations of such pathways are associated with a poorer prognosis, characterized by hospitalization and death [77]. Research is needed to determine if altered miRNA expression is a mechanism interlinking toxic metal exposures with COVID-19 severity and whether there are important windows of susceptibility.
The toxic metal cadmium has also been hypothesized to contribute to COVID-19 outcomes. Cadmium is a major constituent of tobacco smoke and smokers have a markedly increased risk of hospital admission and death from COVID-19 [78, 79]. Although cadmium exposure has not yet been investigated as a mediator of associations between smoking and COVID-19 outcomes, there are data linking cadmium to other relevant endpoints. A 2020 analysis of the National Health and Nutrition Examination Survey (NHANES) participants aged 45 and older found that greater cadmium exposures, as measured in urine and blood, were prospectively associated greater mortality from influenza and pneumonia [80]. Mechanistically, cadmium exposure can modify DNA methylation signatures in whole blood and alter miRNA expression in peripheral blood leukocytes as well as serum [81,82,83]. Of particular interest is a 2021 study in which men occupationally exposed to cadmium were found to have threefold higher expression of miR-221, which was correlated with higher proportions of T helper 17 (Th17) cells and serum IL-17 levels [83]. Th17 cells are a distinct subset of T helper cells that produce the highly pro-inflammatory cytokine IL-17 [84]. Recently, the IL-17 signaling pathway has been shown to become highly activated by SARS-CoV-2 [85], prompting calls to consider it as a possible target for the treatment of COVID-19 [86]. If substantiated by longitudinal data, the data presented here suggest that reducing cadmium exposure and/or epigenetic perturbations could be additional targets for improving COVID-19 outcomes.
Finally, there is evidence that mercury is toxic to humans with wide-ranging effects on the immune system. Methylmercury is the predominant mercury compound encountered in the environment and ingestion of contaminated seafood is the most common route and source of exposure [87]. In an epidemiologic study of fish consumers from the Amazonian region of Brazil, greater methylmercury exposures were associated with higher levels of pro-inflammatory (IL-6, IL-17, and IFN-γ) and anti-inflammatory (IL-4) serum cytokines [88]. That methylmercury was positively associated with both pro- and anti-inflammatory cytokines supports the notion that exposures induce broad immunotoxic effects. Suppression of the developing immune system appears to be one such consequence of prenatal mercury exposure, as evidenced by data from the New Hampshire Birth Cohort Study showing greater maternal mercury exposures during pregnancy from fish consumption (reflecting methylmercury) and having dental amalgams (reflecting elemental mercury) were associated with elevated risks of respiratory infections in infants [89]. In another analysis of New Hampshire Birth Cohort Study data, prenatal mercury exposures were associated with differential DNA methylation signatures in umbilical cord blood that were related to shifts in leukocyte composition [90]. Specifically, cord blood DNA methylation measurements were used to infer leukocyte proportions and greater prenatal mercury exposures were found to correlate with lower estimated proportions of monocytes [90]. Monocytes are a type of leukocyte responsible for identifying and destroying pathogens and eliminating infected cells and individuals with persistently low numbers of monocytes (“monocytopenia”) are at increased risk of developing severe COVID-19 [91]. Whether the pleiotropic effects of mercury are relevant to the development and progression of COVID-19 remains to be studied, but warrants consideration given the ubiquity of mercury in the environment.
Per- and Polyfluorinated Substances (PFAS)
PFAS refer to a group of > 4000 toxic chemicals used in a variety of manufacturing processes, many of which have become environmental contaminants [92]. Within the broad class of PFAS, there are numerous chemicals, including perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), perfluorononanoate (PFNA), perfluorodecanoate (PFDA), perfluorobutanoic acid (PFBA), perfluorohexane sulfonate (PFHxS), and “alternatives” such as GenX [92,93,94]. PFAS are known to accumulate within the body and lead to a variety of outcomes, including immunosuppression, reduced vaccine response, and epigenetic changes [95, 96••]. Recent research has also shown associations between PFAS exposure and COVID-19 severity and mortality [97••, 98]. In particular, elevated plasma concentrations of PFBA have been associated with severe COVID-19 (odds ratio 2.10 [95% CI, 1.02, 4.33]) and strongly associated with ICU admission or death (odds ratio 5.18 [95% CI, 1.29, 20.72]), with effects similarly observed both men and women [97••].
In relation to modifying viral entry, PFAS are suspected to increase ACE2 and TMPRSS2 expression through hypomethylation, leading to increased vulnerability to the SARS-CoV-2 virus [95]. Animal studies support this hypothesis: researchers have observed downregulation of DNA methylation regulators (DNA methyltransferases and Ten Eleven Translocation (TET) enzymes) and upregulated expression of ACE2 and TMPRSS2 in mice following PFOA exposure, in addition to differential methylation in the TMPRSS2 gene promoter [95].
PFAS are also known to alter innate and adaptive immune function, with most studies suggesting suppressive effects [99,100,101,102,103]. For instance, in mouse models, PFAS exposures have been shown to decrease IgM antibody production and levels, a comprehensive measure of general immune response [104, 105]. Other research has associated PFAS with perturbations of T cells [106, 107], B cells [105], macrophages [103], and neutrophils [103], along with atrophy of the thymus and spleen [102]. Alteration of these immune functions may have implications for COVID-19 progression, as delayed or suppressed immune responses are hypothesized to influence disease severity [13••]. However, existing research examining immunosuppression prior to infection has found no significant association with the onset of critical COVID-19 symptoms [108, 109]. Nevertheless, previous studies have been limited due to small sample sizes [108, 109], and PFAS-associated immunosuppression may still predispose an individual to more severe COVID-19 outcomes. In addition to immune suppression, PFAS have been associated with dysregulation of inflammatory responses and chronic inflammation in humans, pathways known to play a role in COVID-19 development [101, 110]. Specific outcomes and pathways vary by study type (rodent, cell culture, epidemiological, etc.), but have included peroxisome proliferator-activated receptor alpha (PPAR-α), NF-kB, IFNs, and various cytokines [101].
It is plausible that some of the immunomodulatory effects associated with PFAS may be under epigenetic regulation. In a 2021 study of firefighters, exposures to PFOA, PFOS, PFNA, PFDA, and other PFAS were associated with changes in DNA methylation for a number of genes associated with immune function [96••]. In particular, PFNA was associated with differential methylation of the Schlafen family member 12 (SLFN12) and IL-32 genes that are upregulated during viral infection and T cell activation [111, 112], while PFDA was associated with differential methylation of dual specificity phosphatase 19 (DUSP19) and glycerol-3-phosphate dehydrogenase 2 (GPD2), two genes involved in regulating inflammatory response [96••, 113, 114]. Other research has shown hypermethylation following PFOS exposures of the genes ring finger protein 39 (RNF39) and major histocompatibility complex, class II, DQ beta 1 (HLA-DQB1), both associated with autoimmune conditions [115, 116]. Taken together, these studies provide evidence that PFAS may alter COVID-19 risk via similar epigenetically-regulated immune pathways; further research is necessary to investigate this relationship.
Finally, PFAS are associated with decrease effectiveness of various vaccines, including tetanus [117, 118], diphtheria [118, 119], measles [120], rubella [121], and influenza [122], particularly when immunized following PFAS exposures in early life. Because of this, concerns have been raised that PFAS may reduce COVID-19 vaccine effectiveness, potentially increasing susceptibility to breakthrough infections or temporally shortening the duration of protection. Although no known research has explored the intersection of PFAS, vaccine effectiveness, and epigenetic alterations, the epigenetic immune modifications previously discussed may provide mechanistic insights into the decreased antibody response observed after vaccination.
Other Endocrine Disrupting Chemicals (EDCs)
The broad class of EDCs represent contaminants that alter normative hormone function in the body, resulting in vast complications. In addition to metals and PFAS (each discussed above), EDCs include bisphenol A (BPA), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), flame retardants, pesticides, phthalates, polychlorinated and polybrominated biphenyls (PCBs and PBBs), polycyclic aromatic hydrocarbons (PAHs), dichlorodiphenyltrichloroethane (DDT), and hundreds more [123]. Not surprisingly, EDC exposure has been suggested to increase COVID-19 risk through a variety of immune pathways [124••, 125, 126]; this section will consider a few examples, but there are hundreds of EDCs that could be discussed [127].
The entry of SARS-CoV-2 into the body depends, at least in part, on the expression of ACE2 and TMPRSS2. Altered expression of these genes following EDC exposure, specifically BPA, has been observed in some, but not all studies [19••, 126, 128]. While more research is necessary to determine a consistent association, complex epigenetic regulation may be involved in altering expression based on known epigenetic involvement with ACE2/TMPRSS2 [41] and EDCs [127].
EDCs are also known to suppress the immune system, potentially conferring COVID-19 risk. TCDD, for example, is known as a potent immunosuppressant, altering the aryl hydrocarbon receptor [129] and resulting in dysregulated T cell differentiation [130•]. TCDD-associated epigenetic regulation of FOXP3 and IL-17 is understood as a part of this immunosuppressive pathway, altering the master regulator of regulatory T cells [130•, 131]. As previously discussed, a weak initial immune response to the SARS-CoV-2 virus may predispose an individual to a more severe course of disease.
EDCs additionally dysregulate numerous facets of the immune system, including T cells, natural killer cells, dendritic cells, macrophages, and neutrophils [127, 132, 133]. For example, BPA has been shown to interact with cell signaling pathways, dysregulating the innate and adaptive immune system by altering chemokine/cytokine levels (observed in COVID-19 patients), inducing inflammation and related inflammatory diseases [126, 133, 134], and altering the function of numerous immune cells [133]. TCDD has shown similar dysregulation of adaptive immune responses, altering T cell function and differentiation [129]. These selected examples are not unique; other EDCs have demonstrated similar effects that alter immune function [135, 136]. Immunomodulation by BPA, TCDD, and other EDCs may exacerbate COVID-19 immune dysregulation, increasing susceptibility for severe disease.
Epigenetic dysregulation is a proposed mechanism by which EDCs may lead to biological effects, both from directly interacting with the epigenome and from perturbing hormonal pathways that alter the epigenome [127]. EDC-associated epigenetic changes in DNA methylation levels, miRNAs, and histone acetylation have been observed related to the immune system for TCDD [137], BPA [138], DDT [138], PCBs [139], PBBs [140], nonylphenol [141], and 4-octylphenol [141], with many EDCs understudied. Some of these epigenetic changes have been linked to transgenerational health effects, increasing the complexity and importance of EDC exposure [142]. We suggest the examination of epigenetically-regulated immune pathways as a recommendation for future study investigating EDC exposure and COVID-19 susceptibility.
Potential Implications and Further Study
While direct evidence that environmental chemicals contribute to the current pandemic is still sparse, it is clear that exposures to such chemicals may impact host susceptibility to SARS-CoV-2 and the clinical course of COVID-19. Given the breadth of chemicals in the environment and their known impacts on human health, there are several ways to move forward. Most broadly, there is a need for continued public and private environmental cleanup efforts to reduce human exposures to toxic chemicals, mitigating some of the effects detailed above. In relation to COVID-19, there is a need to weigh environmental contaminant exposures as risk factors and to prioritize the following research:
-
1)
Quantify the direct impacts of exposures to air pollutants, toxic metal(loid)s, per- and polyfluorinated substances, other endocrine disrupting chemicals, and their mixtures on COVID-19 susceptibility and severity.
-
2)
Understand the specific epigenetic mechanisms and immune pathways linking environmental chemical exposures and adverse COVID-19 outcomes, especially whether any are reversible.
-
3)
Identify susceptible widows of exposure and sensitive sub-groups, including assessments of early life exposures and differences in exposure-outcome associations by biological sex.
-
4)
Discover and validate minimally-invasive biomarkers that can identify individuals at risk of severe COVID-19 due to environmental chemical exposures.
-
5)
Develop interventions to limit chemicals in the environment or mitigate adverse health impacts of exposures in severe COVID-19 cases (e.g., for metal-associated immunotoxicity, interventions could include nutritional supplementation or chelation therapy).
-
6)
Evaluate the need for boosters to strengthen COVID-19 vaccine effectiveness among individuals highly exposed to immunotoxic environmental chemicals.
-
7)
Converge aspects of epidemiologic and experimental study design and dissemination of findings that can support rapid environmental policy making.
Finally, greater collaboration between environmental health scientists, molecular epidemiologists, immunologists, and infectious disease researchers conducting basic and translational science is needed to promote increased understanding of the interactions between environmental chemicals and infectious diseases, including epigenetic contributions. Breaking down disciplinary silos would better enable taking adaptive actions to respond to COVID-19 and pre-emptive actions to possibly prevent future pandemics.
Conclusion
In conclusion, numerous environmental contaminants are associated with epigenetic modification of immunomodulatory genes involved in viral entry, viral recognition, cytokine production, and immunologic memory. Several of these immune pathways overlap with those involved in the host response to the SARS-CoV-2 virus. Epigenetic regulation of the immune system may be a significant molecular mechanism underpinning associations between environmental chemicals and COVID-19 outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Escandón K, Rasmussen AL, Bogoch II, Murray EJ, Escandón K, Popescu SV, et al. COVID-19 false dichotomies and a comprehensive review of the evidence regarding public health, COVID-19 symptomatology, SARS-CoV-2 transmission, mask wearing, and reinfection. BMC Infect Dis. 2021;21(1):710. https://doi.org/10.1186/s12879-021-06357-4.
Self WH, Tenforde MW, Rhoads JP, Gaglani M, Ginde AA, Douin DJ, et al. Comparative effectiveness of Moderna, Pfizer-BioNTech, and Janssen (Johnson & Johnson) vaccines in preventing COVID-19 hospitalizations among adults without immunocompromising conditions - United States, March-August 2021. MMWR Morb Mortal Wkly Rep. 2021;70(38):1337–43. https://doi.org/10.15585/mmwr.mm7038e1.
Barek MA, Aziz MA, Islam MS. Impact of age, sex, comorbidities and clinical symptoms on the severity of COVID-19 cases: A meta-analysis with 55 studies and 10014 cases. Heliyon. 2020;6(12):e05684. https://doi.org/10.1016/j.heliyon.2020.e05684.
Liu W, Yang C, Liao Y-g, Wan F, Lin L, Huang X, et al. Risk factors for COVID-19 progression and mortality in hospitalized patients without pre-existing comorbidities. J Infect Public Health. 2022;15(1):13–20. https://doi.org/10.1016/j.jiph.2021.11.012.
Wolff D, Nee S, Hickey NS, Marschollek M. Risk factors for Covid-19 severity and fatality: a structured literature review. Infection. 2021;49(1):15–28. https://doi.org/10.1007/s15010-020-01509-1.
•• Travaglio M, Yu Y, Popovic R, Selley L, Leal NS, Martins LM. Links between air pollution and COVID-19 in England. Environ Pollut. 2021;268:115859. https://doi.org/10.1016/j.envpol.2020.115859. This study combined regional- and individual-level data to quantify associations of air pollutants with COVID-19 incidence and mortality.
Germolec D, Luebke R, Rooney A, Shipkowski K, Vandebriel R, van Loveren H. Immunotoxicology: A brief history, current status and strategies for future immunotoxicity assessment. Curr Opin Toxicol. 2017;5:55–9. https://doi.org/10.1016/j.cotox.2017.08.002.
World Health Organization. Principles and Methods for Assessing Direct Immunotoxicity Associated with Exposure to Chemicals-Environmental Health Criteria 180. 1996.
National Academies of Sciences E, Medicine. Toward Understanding the Interplay of Environmental Stressors, Infectious Diseases, and Human Health: Proceedings of a Workshop—in Brief. Washington, DC: The National Academies Press; 2019.
World Health Organization. State of the Science of Endocrine Disrupting Chemicals. World Health Organization; 2012.
World Health Organization: IARC Monographs on the Identification of Carcinogenic Hazards to Humans. https://monographs.iarc.who.int/monographs-available/ (2012). Accessed 3/2/22.
•• Paludan SR, Mogensen TH. Innate immunological pathways in COVID-19 pathogenesis. Sci Immunol. 2022;7(67):eamb5505. https://doi.org/10.1126/sciimmunol.abm5505. This manuscript provides a background of the innate immune response to COVID-19, which is necessary to understanding the impacts of environmental chemical exposures and the contribution of epigenetic mechanisms.
•• Sette A, Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184(4):861–80. https://doi.org/10.1016/j.cell.2021.01.007. This manuscript provides a background of the other arm of the immune system, adaptive immunity, in response to COVID-19, which is also necessary to understanding the impacts of environmental chemical exposures and contributions of epigenetic mechanisms.
Bonnet B, Cosme J, Dupuis C, Coupez E, Adda M, Calvet L, et al. Severe COVID-19 is characterized by the co-occurrence of moderate cytokine inflammation and severe monocyte dysregulation. EBioMedicine. 2021;73:103622. https://doi.org/10.1016/j.ebiom.2021.103622.
Wu H, He P, Ren Y, Xiao S, Wang W, Liu Z, et al. Postmortem high-dimensional immune profiling of severe COVID-19 patients reveals distinct patterns of immunosuppression and immunoactivation. Nat Commun. 2022;13(1):269. https://doi.org/10.1038/s41467-021-27723-5.
Loftus TJ, Ungaro R, Dirain M, Efron PA, Mazer MB, Remy KE, et al. Overlapping but disparate inflammatory and immunosuppressive responses to SARS-CoV-2 and Bacterial Sepsis: An immunological time course analysis. Front Immunol. 2021;12:792448. https://doi.org/10.3389/fimmu.2021.792448.
Bhalla V, Blish CA, South AM. A historical perspective on ACE2 in the COVID-19 era. J Hum Hypertens. 2021;35(10):935–9. https://doi.org/10.1038/s41371-020-00459-3.
Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24(1):1–10.
•• Rath S, Perikala V, Jena AB, Dandapat J. Factors regulating dynamics of angiotensin-converting enzyme-2 (ACE2), the gateway of SARS-CoV-2: Epigenetic modifications and therapeutic interventions by epidrugs. Biomed Pharmacother. 2021;143:112095. https://doi.org/10.1016/j.biopha.2021.112095. This review emphasizes the relevance of environmental contaminants and epigenetic regulation to COVID-19 susceptibility via the ACE2 receptor.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271-80.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Andolfo I, Russo R, Lasorsa VA, Cantalupo S, Rosato BE, Bonfiglio F, et al. Common variants at 21q22.3 locus influence MX1 and TMPRSS2 gene expression and susceptibility to severe COVID-19. iScience. 2021;24(4):102322. https://doi.org/10.1016/j.isci.2021.102322.
Xu G, Qi F, Wang H, Liu Y, Wang X, Zou R, et al. The transient IFN response and the delay of adaptive immunity feature the severity of COVID-19. Front Immunol. 2022;12:816745. https://doi.org/10.3389/fimmu.2021.816745.
Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. 2021;6(1):255. https://doi.org/10.1038/s41392-021-00679-0.
Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184(1):149-68.e17. https://doi.org/10.1016/j.cell.2020.11.025.
Schultze JL, Aschenbrenner AC. COVID-19 and the human innate immune system. Cell. 2021;184(7):1671–92. https://doi.org/10.1016/j.cell.2021.02.029.
Kalicińska E, Szymczak D, Zińczuk A, Adamik B, Smiechowicz J, Skalec T, et al. Immunosuppression as a hallmark of critical COVID-19: prospective study. Cells. 2021;10(6):1293.
Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight. 2020;5(17):e140329. https://doi.org/10.1172/jci.insight.140329.
Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–9. https://doi.org/10.1038/s41587-020-0602-4.
Isho B, Abe KT, Zuo M, Jamal AJ, Rathod B, Wang JH, et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol. 2020;5(52). https://doi.org/10.1126/sciimmunol.abe5511.
Painter MM, Mathew D, Goel RR, Apostolidis SA, Pattekar A, Kuthuru O, et al. Rapid induction of antigen-specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity. 2021;54(9):2133-42.e3. https://doi.org/10.1016/j.immuni.2021.08.001.
COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature. 2021;600(7889):472–7. https://doi.org/10.1038/s41586-021-03767-x.
•• Konigsberg IR, Barnes B, Campbell M, Davidson E, Zhen Y, Pallisard O, et al. Host methylation predicts SARS-CoV-2 infection and clinical outcome. Commun Med (London). 2021;1(1):42. https://doi.org/10.1038/s43856-021-00042-y. This study establishes DNA methylation of immune-related genes as predictive of COVID-19 infection and severity, showing individual susceptibility and responses to infection are under epigenetic control.
•• Castro de Moura M, Davalos V, Planas-Serra L, Alvarez-Errico D, Arribas C, Ruiz M, et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine. 2021;66:103339. https://doi.org/10.1016/j.ebiom.2021.103339. This study establishes DNA methylation of immune-related genes as predictive of COVID-19 outcomes, emphasizing the role of epigenetic regulation in disease prognosis.
Zhang G, Pradhan S. Mammalian epigenetic mechanisms. IUBMB Life. 2014;66(4):240–56. https://doi.org/10.1002/iub.1264.
Ogen Y. Assessing nitrogen dioxide (NO2) levels as a contributing factor to coronavirus (COVID-19) fatality. Sci Total Environ. 2020;726:138605.
Lai A, Chang ML, O’Donnell RP, Zhou C, Sumner JA, Hsiai TK. Association of COVID-19 transmission with high levels of ambient pollutants: Initiation and impact of the inflammatory response on cardiopulmonary disease. Sci Total Environ. 2021;779:146464.
Wu X, Nethery RC, Sabath MB, Braun D, Dominici F. Exposure to air pollution and COVID-19 mortality in the United States: A nationwide cross-sectional study. 2020.
Gupta A, Bherwani H, Gautam S, Anjum S, Musugu K, Kumar N, et al. Air pollution aggravating COVID-19 lethality? Exploration in Asian cities using statistical models. Environ Dev Sustain. 2021;23(4):6408–17.
Conticini E, Frediani B, Caro D. Can atmospheric pollution be considered a co-factor in extremely high level of SARS-CoV-2 lethality in Northern Italy? Environ Pollut. 2020;261:114465.
Sagawa T, Tsujikawa T, Honda A, Miyasaka N, Tanaka M, Kida T, et al. Exposure to particulate matter upregulates ACE2 and TMPRSS2 expression in the murine lung. Environ Res. 2021;195:110722. https://doi.org/10.1016/j.envres.2021.110722.
Beacon TH, Delcuve GP, Davie JR. Epigenetic regulation of ACE2, the receptor of the SARS-CoV-2 virus1. Genome. 2021;64(4):386–99.
Paniri A, Hosseini MM, Akhavan-Niaki H. First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J Biomol Struct Dyn. 2021;39(10):3576–93. https://doi.org/10.1080/07391102.2020.1767690.
• Cardenas A, Rifas-Shiman SL, Sordillo JE, DeMeo DL, Baccarelli AA, Hivert MF, et al. DNA methylation architecture of the ACE2 gene in nasal cells of children. Sci Rep. 2021;11(1):7107. https://doi.org/10.1038/s41598-021-86494-7. This study demonstrates methylation of ACE2, the receptor through which SARS-CoV-2 gains entry into nasal cells that are in contact with air pollutants, differs by demographic characteristics including age, sex, and race.
Alfano R, Herceg Z, Nawrot TS, Chadeau-Hyam M, Ghantous A, Plusquin M. The impact of air pollution on our epigenome: how far is the evidence?(A systematic review). Curr Environ Health Rep. 2018;5(4):544–78.
Møller P, Danielsen PH, Karottki DG, Jantzen K, Roursgaard M, Klingberg H, et al. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat Res Rev Mutat Res. 2014;762:133–66. https://doi.org/10.1016/j.mrrev.2014.09.001.
Paulin L, Hansel N. Particulate air pollution and impaired lung function. F1000Res. 2016;5:F1000 Faculty Rev-201. https://doi.org/10.12688/f1000research.7108.1.
Ciencewicki J, Jaspers I. Air pollution and respiratory viral infection. Inhalation Toxicol. 2007;19(14):1135–46.
Signorini C, Pignatti P, Coccini T. How do inflammatory mediators, immune response and air pollution contribute to COVID-19 disease severity? A lesson to learn. Life. 2021;11(3):182.
Plummer LE, Smiley-Jewell S, Pinkerton KE. Impact of air pollution on lung inflammation and the role of Toll-like receptors. Int J Interf Cytokine Mediator Res. 2012;4:43–57.
Zhong J, Colicino E, Lin X, Mehta A, Kloog I, Zanobetti A, et al. Cardiac autonomic dysfunction: particulate air pollution effects are modulated by epigenetic immunoregulation of toll-like receptor 2 and dietary flavonoid intake. J Am Heart Assoc. 2015;4(1):e001423.
Bellavia A, Urch B, Speck M, Brook RD, Scott JA, Albetti B, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2(3):e000212.
Kohli A, Garcia MA, Miller RL, Maher C, Humblet O, Hammond SK, et al. Secondhand smoke in combination with ambient air pollution exposure is associated with increasedx CpG methylation and decreased expression of IFN-γ in T effector cells and Foxp3 in T regulatory cells in children. Clin Epigenetics. 2012;4(1):17. https://doi.org/10.1186/1868-7083-4-17.
Bind M-A, Lepeule J, Zanobetti A, Gasparrini A, Baccarelli AA, Coull BA, et al. Air pollution and gene-specific methylation in the Normative Aging Study. Epigenetics. 2014;9(3):448–58. https://doi.org/10.4161/epi.27584.
Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol. 2010;126(4):845-52.e10. https://doi.org/10.1016/j.jaci.2010.08.008.
Zhang S, Chen S, Xiao G, Zhao M, Li J, Dong W, et al. The associations between air pollutant exposure and neutralizing antibody titers of an inactivated SARS-CoV-2 vaccine. Environ Sci Pollut Res. 2022;29(9):13720–8. https://doi.org/10.1007/s11356-021-16786-y.
Franza L, Cianci R. Pollution, Inflammation, and Vaccines: A Complex Crosstalk. Int J Environ Res Public Health. 2021;18(12):6330.
Agency for Toxic Substances and Disease Registry: Substance Priority List. https://www.atsdr.cdc.gov/spl/index.html (2019). Accessed.
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ. Heavy metal toxicity and the environment. Exp Suppl. 2012;101:133–64. https://doi.org/10.1007/978-3-7643-8340-4_6.
Krueger WS, Wade TJ. Elevated blood lead and cadmium levels associated with chronic infections among non-smokers in a cross-sectional analysis of NHANES data. Environ Health. 2016;15:16. https://doi.org/10.1186/s12940-016-0113-4.
Park WJ, Kim SH, Kang W, Ahn JS, Cho S, Lim DY, et al. Blood lead level and Helicobacter pylori infection in a healthy population: A cross-sectional study. Arch Environ Occup Health. 2020;75(6):333–8. https://doi.org/10.1080/19338244.2019.1654969.
Sirivarasai J, Wananukul W, Kaojarern S, Chanprasertyothin S, Thongmung N, Ratanachaiwong W, et al. Association between inflammatory marker, environmental lead exposure, and glutathione S-transferase gene. Biomed Res Int. 2013;2013:474963. https://doi.org/10.1155/2013/474963.
Kim JH, Lee KH, Yoo DH, Kang D, Cho SH, Hong YC. GSTM1 and TNF-alpha gene polymorphisms and relations between blood lead and inflammatory markers in a non-occupational population. Mutat Res. 2007;629(1):32–9. https://doi.org/10.1016/j.mrgentox.2007.01.004.
Xu X, Chen X, Zhang J, Guo P, Fu T, Dai Y, et al. Decreased blood hepatitis B surface antibody levels linked to e-waste lead exposure in preschool children. J Hazard Mater. 2015;298:122–8. https://doi.org/10.1016/j.jhazmat.2015.05.020.
Conterato GM, Bulcão RP, Sobieski R, Moro AM, Charão MF, de Freitas FA, et al. Blood thioredoxin reductase activity, oxidative stress and hematological parameters in painters and battery workers: relationship with lead and cadmium levels in blood. J Appl Toxicol. 2013;33(2):142–50. https://doi.org/10.1002/jat.1731.
Hsiao CL, Wu KH, Wan KS. Effects of environmental lead exposure on T-helper cell-specific cytokines in children. J Immunotoxicol. 2011;8(4):284–7. https://doi.org/10.3109/1547691x.2011.592162.
Karmaus W, Brooks KR, Nebe T, Witten J, Obi-Osius N, Kruse H. Immune function biomarkers in children exposed to lead and organochlorine compounds: a cross-sectional study. Environ Health. 2005;4(1):5. https://doi.org/10.1186/1476-069x-4-5.
Boscolo P, Di Gioacchino M, Sabbioni E, Di Giacomo F, Reale M, Volpe AR, et al. Lymphocyte subpopulations, cytokines and trace elements in asymptomatic atopic women exposed to an urban environment. Life Sci. 2000;67(10):1119–26. https://doi.org/10.1016/s0024-3205(00)00712-8.
• Zeng HL, Zhang B, Wang X, Yang Q, Cheng L. Urinary trace elements in association with disease severity and outcome in patients with COVID-19. Environ Res. 2021;194:110670. https://doi.org/10.1016/j.envres.2020.110670. This study establishes associations between several urinary toxic metal(loid) levels and poor COVID-19 outcomes.
Sen A, Heredia N, Senut MC, Hess M, Land S, Qu W, et al. Early life lead exposure causes gender-specific changes in the DNA methylation profile of DNA extracted from dried blood spots. Epigenomics. 2015;7(3):379–93. https://doi.org/10.2217/epi.15.2.
Mevel R, Draper JE, Lie ALM, Kouskoff V, Lacaud G. RUNX transcription factors: orchestrators of development. Development. 2019;146(17). https://doi.org/10.1242/dev.148296.
Oja AE, Saris A, Ghandour CA, Kragten NAM, Hogema BM, Nossent EJ, et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur J Immunol. 2020;50(12):1998–2012. https://doi.org/10.1002/eji.202048908.
Gomez JMD, Du-Fay-de-Lavallaz JM, Fugar S, Sarau A, Simmons JA, Clark B, et al. Sex differences in COVID-19 hospitalization and mortality. J Womens Health (Larchmt). 2021;30(5):646–53. https://doi.org/10.1089/jwh.2020.8948.
Farzan SF, Li Z, Korrick SA, Spiegelman D, Enelow R, Nadeau K, et al. Infant infections and respiratory symptoms in relation to in utero arsenic exposure in a U.S. cohort. Environ Health Perspect. 2016;124(6):840–7. https://doi.org/10.1289/ehp.1409282.
Rahman A, Vahter M, Ekström EC, Persson L. Arsenic exposure in pregnancy increases the risk of lower respiratory tract infection and diarrhea during infancy in Bangladesh. Environ Health Perspect. 2011;119(5):719–24. https://doi.org/10.1289/ehp.1002265.
•• Hsu KS, Goodale BC, Ely KH, Hampton TH, Stanton BA, Enelow RI. Single-cell RNA-seq analysis reveals that prenatal arsenic exposure results in long-term, adverse effects on immune gene expression in response to influenza A infection. Toxicol Sci. 2020;176(2):312–28. https://doi.org/10.1093/toxsci/kfaa080. This manuscript shows that in utero arsenic exposure is associated with greater lung damage and inflammation following influenza virus infection, stemming from a dysregulated immune response.
Rager JE, Bailey KA, Smeester L, Miller SK, Parker JS, Laine JE, et al. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environ Mol Mutagen. 2014;55(3):196–208. https://doi.org/10.1002/em.21842.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–4. https://doi.org/10.1016/s0140-6736(20)30628-0.
Caruso RV, O’Connor RJ, Stephens WE, Cummings KM, Fong GT. Toxic metal concentrations in cigarettes obtained from U.S. smokers in 2009: results from the International Tobacco Control (ITC) United States survey cohort. Int J Environ Res Public Health. 2013;11(1):202–17. https://doi.org/10.3390/ijerph110100202.
Lowe KE, Zein J, Hatipoglu U, Attaway A. Association of smoking and cumulative pack-year exposure with COVID-19 outcomes in the Cleveland Clinic COVID-19 registry. JAMA Intern Med. 2021;181(5):709–11. https://doi.org/10.1001/jamainternmed.2020.8360.
Park SK, Sack C, Sirén MJ, Hu H. Environmental cadmium and mortality from influenza and pneumonia in U.S. adults. Environ Health Perspect. 2020;128(12):127004. https://doi.org/10.1289/ehp7598.
Domingo-Relloso A, Riffo-Campos AL, Haack K, Rentero-Garrido P, Ladd-Acosta C, Fallin DM, et al. Cadmium, smoking, and human blood DNA methylation profiles in adults from the strong heart study. Environ Health Perspect. 2020;128(6):67005. https://doi.org/10.1289/ehp6345.
Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ Health Perspect. 2010;118(6):763–8. https://doi.org/10.1289/ehp.0901300.
Goyal T, Mitra P, Singh P, Ghosh R, Sharma S, Sharma P. Association of microRNA expression with changes in immune markers in workers with cadmium exposure. Chemosphere. 2021;274:129615. https://doi.org/10.1016/j.chemosphere.2021.129615.
Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8(4):345–50. https://doi.org/10.1038/ni0407-345.
Hasan MZ, Islam S, Matsumoto K, Kawai T. SARS-CoV-2 infection initiates interleukin-17-enriched transcriptional response in different cells from multiple organs. Sci Rep. 2021;11(1):16814. https://doi.org/10.1038/s41598-021-96110-3.
Pacha O, Sallman MA, Evans SE. COVID-19: a case for inhibiting IL-17? Nat Rev Immunol. 2020;20(6):345–6. https://doi.org/10.1038/s41577-020-0328-z.
Agency for Toxic Substances and Disease Registry. Toxicological Profile for Mercury. In: U.S. Department of Health and Human Services, editor. Atlanta, GA; 1999.
Nyland JF, Fillion M, Barbosa F Jr, Shirley DL, Chine C, Lemire M, et al. Biomarkers of methylmercury exposure immunotoxicity among fish consumers in Amazonian Brazil. Environ Health Perspect. 2011;119(12):1733–8. https://doi.org/10.1289/ehp.1103741.
Emeny RT, Korrick SA, Li Z, Nadeau K, Madan J, Jackson B, et al. Prenatal exposure to mercury in relation to infant infections and respiratory symptoms in the New Hampshire Birth Cohort Study. Environ Res. 2019;171:523–9. https://doi.org/10.1016/j.envres.2019.01.026.
Cardenas A, Koestler DC, Houseman EA, Jackson BP, Kile ML, Karagas MR, et al. Differential DNA methylation in umbilical cord blood of infants exposed to mercury and arsenic in utero. Epigenetics. 2015;10(6):508–15. https://doi.org/10.1080/15592294.2015.1046026.
Pehlivan O, Serin SO, Yesil EE, Demirdag F. Can Monocytopenia Be a New Indicator in Determining Survival in Covid-19 Disease? Clin Lab. 2021;67(10). https://doi.org/10.7754/Clin.Lab.2021.210215.
OECD. Toward a new comprehensive global database of per-and polyfluoroalkyl substances (PFASs): Summary report on updating the OECD 2007 list of per-and polyfluoroalkyl substances (PFASs). Organisation for Economic Cooperation and Development (OECD). 2018.
Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag. 2011;7(4):513–41. https://doi.org/10.1002/ieam.258.
Hopkins ZR, Sun M, DeWitt JC, Knappe DR. Recently detected drinking water contaminants: GenX and other per-and polyfluoroalkyl ether acids. J Am Water Works Ass. 2018;110(7):13–28.
Ahmad S, Wen Y, Irudayaraj JMK. PFOA induces alteration in DNA methylation regulators and SARS-CoV-2 targets Ace2 and Tmprss2 in mouse lung tissues. Toxicol Rep. 2021;8:1892–8. https://doi.org/10.1016/j.toxrep.2021.11.014.
•• Goodrich JM, Calkins MM, Caban-Martinez AJ, Stueckle T, Grant C, Calafat AM, et al. Per-and polyfluoroalkyl substances, epigenetic age and DNA methylation: a cross-sectional study of firefighters. Epigenomics. 2021;13(20):1619–36. This study of firefighters shows methylation of immune-related genes differs by levels of PFAS exposure.
•• Grandjean P, Timmermann CAG, Kruse M, Nielsen F, Vinholt PJ, Boding L, et al. Severity of COVID-19 at elevated exposure to perfluorinated alkylates. PLOS ONE. 2021;15(12):e0244815. https://doi.org/10.1371/journal.pone.0244815. This manuscript was the first to identify strong associations between PFAS exposures and COVID-19 outcomes in an epidemiologic cohort, highlighting the importance of investigating environmental chemical exposures in the context of the pandemic.
Catelan D, Biggeri A, Russo F, Gregori D, Pitter G, Da Re F, et al. Exposure to perfluoroalkyl substances and mortality for COVID-19: A spatial ecological analysis in the Veneto Region (Italy). Int J Environ Res Public Health. 2021;18(5):2734.
Pinder M, Fry RC, Alexis NE. Chapter 9 - Environmental contaminants and the immune system: A systems perspective. In: Fry RC, editor. Environmental Epigenetics in Toxicology and Public Health. Academic Press; 2020. p. 217–37.
Sly PD, Trottier BA, Bulka CM, Cormier SA, Fobil J, Fry RC, et al. The interplay between environmental exposures and COVID-19 risks in the health of children. Environ Health. 2021;20(1):34. https://doi.org/10.1186/s12940-021-00716-z.
DeWitt JC, Peden-Adams MM, Keller JM, Germolec DR. Immunotoxicity of perfluorinated compounds: recent developments. Toxicol Pathol. 2012;40(2):300–11. https://doi.org/10.1177/0192623311428473.
Qazi MR, Xia Z, Bogdanska J, Chang S-C, Ehresman DJ, Butenhoff JL, et al. The atrophy and changes in the cellular compositions of the thymus and spleen observed in mice subjected to short-term exposure to perfluorooctanesulfonate are high-dose phenomena mediated in part by peroxisome proliferator-activated receptor-alpha (PPARα). Toxicology. 2009;260(1–3):68–76.
Qazi MR, Bogdanska J, Butenhoff JL, Nelson BD, DePierre JW, Abedi-Valugerdi M. High-dose, short-term exposure of mice to perfluorooctanesulfonate (PFOS) or perfluorooctanoate (PFOA) affects the number of circulating neutrophils differently, but enhances the inflammatory responses of macrophages to lipopolysaccharide (LPS) in a similar fashion. Toxicology. 2009;262(3):207–14. https://doi.org/10.1016/j.tox.2009.06.010.
Dewitt JC, Copeland CB, Strynar MJ, Luebke RW. Perfluorooctanoic acid-induced immunomodulation in adult C57BL/6J or C57BL/6N female mice. Environ Health Perspect. 2008;116(5):644–50. https://doi.org/10.1289/ehp.10896.
Peden-Adams MM, Keller JM, Eudaly JG, Berger J, Gilkeson GS, Keil DE. Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicol Sci. 2008;104(1):144–54. https://doi.org/10.1093/toxsci/kfn059.
Zheng L, Dong G-H, Zhang Y-H, Liang Z-F, Jin Y-H, He Q-C. Type 1 and Type 2 cytokines imbalance in adult male C57BL/6 mice following a 7-day oral exposure to perfluorooctanesulfonate (PFOS). J Immunotoxicol. 2011;8(1):30–8.
Dong G-H, Liu M-M, Wang D, Zheng L, Liang Z-F, Jin Y-H. Sub-chronic effect of perfluorooctanesulfonate (PFOS) on the balance of type 1 and type 2 cytokine in adult C57BL6 mice. Arch Toxicol. 2011;85(10):1235–44.
Gao Y, Chen Y, Liu M, Shi S, Tian J. Impacts of immunosuppression and immunodeficiency on COVID-19: A systematic review and meta-analysis. J Infect. 2020;81(2):e93–5. https://doi.org/10.1016/j.jinf.2020.05.017.
Tassone D, Thompson A, Connell W, Lee T, Ungaro R, An P, et al. Immunosuppression as a risk factor for COVID-19: a meta-analysis. Intern Med J. 2021;51(2):199–205. https://doi.org/10.1111/imj.15142.
Omoike OE, Pack RP, Mamudu HM, Liu Y, Strasser S, Zheng S, et al. Association between per and polyfluoroalkyl substances and markers of inflammation and oxidative stress. Environ Res. 2021;196:110361. https://doi.org/10.1016/j.envres.2020.110361.
Liu F, Zhou P, Wang Q, Zhang M, Li D. The Schlafen family: complex roles in different cell types and virus replication. Cell Biol Int. 2018;42(1):2–8. https://doi.org/10.1002/cbin.10778.
Li W, Sun W, Liu L, Yang F, Li Y, Chen Y, et al. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J Immunol. 2010;185(9):5056–65.
Langston PK, Nambu A, Jung J, Shibata M, Aksoylar HI, Lei J, et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat Immunol. 2019;20(9):1186–95. https://doi.org/10.1038/s41590-019-0453-7.
Xie X-K, Xu Z-K, Xu K, Xiao Y-X. DUSP19 mediates spinal cord injury-induced apoptosis and inflammation in mouse primary microglia cells via the NF-kB signaling pathway. Neurol Res. 2020;42(1):31–8. https://doi.org/10.1080/01616412.2019.1685068.
van den Dungen MW, Murk AJ, Kampman E, Steegenga WT, Kok DE. Association between DNA methylation profiles in leukocytes and serum levels of persistent organic pollutants in Dutch men. Environ Epigenet. 2017;3(1). https://doi.org/10.1093/eep/dvx001.
Maltby VE, Lea RA, Sanders KA, White N, Benton MC, Scott RJ, et al. Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin Epigenetics. 2017;9(1):71. https://doi.org/10.1186/s13148-017-0371-1.
Grandjean P, Heilmann C, Weihe P, Nielsen F, Mogensen UB, Timmermann A, et al. Estimated exposures to perfluorinated compounds in infancy predict attenuated vaccine antibody concentrations at age 5-years. J Immunotoxicol. 2017;14(1):188–95.
Kielsen K, Shamim Z, Ryder LP, Nielsen F, Grandjean P, Budtz-Jørgensen E, et al. Antibody response to booster vaccination with tetanus and diphtheria in adults exposed to perfluorinated alkylates. J Immunotoxicol. 2016;13(2):270–3.
Grandjean P, Heilmann C, Weihe P, Nielsen F, Mogensen UB, Budtz-Jørgensen E. Serum vaccine antibody concentrations in adolescents exposed to perfluorinated compounds. Environ Health Perspect. 2017;125(7):077018. https://doi.org/10.1289/EHP275.
Timmermann CAG, Jensen KJ, Nielsen F, Budtz-Jørgensen E, van der Klis F, Benn CS, et al. Serum perfluoroalkyl substances, vaccine responses, and morbidity in a cohort of Guinea-Bissau children. Environ Health Perspect. 2020;128(8):087002.
Granum B, Haug LS, Namork E, Stølevik SB, Thomsen C, Aaberge IS, et al. Pre-natal exposure to perfluoroalkyl substances may be associated with altered vaccine antibody levels and immune-related health outcomes in early childhood. J Immunotoxicol. 2013;10(4):373–9.
Looker C, Luster MI, Calafat AM, Johnson VJ, Burleson GR, Burleson FG, et al. Influenza Vaccine Response in Adults Exposed to Perfluorooctanoate and Perfluorooctanesulfonate. Toxicol Sci. 2013;138(1):76–88. https://doi.org/10.1093/toxsci/kft269.
Karthikeyan BS, Ravichandran J, Mohanraj K, Vivek-Ananth RP, Samal A. A curated knowledgebase on endocrine disrupting chemicals and their biological systems-level perturbations. Sci Total Environ. 2019;692:281–96. https://doi.org/10.1016/j.scitotenv.2019.07.225.
•• Wu Q, Coumoul X, Grandjean P, Barouki R, Audouze K. Endocrine disrupting chemicals and COVID-19 relationships: A computational systems biology approach. Environ Int. 2021;157:106232. https://doi.org/10.1016/j.envint.2020.106232. This manuscript provides initial evidence from using an in silico approach that exposures to endocrine disrupting chemicals may disrupt signaling pathways important for COVID-19 severity.
Tsatsakis A, Petrakis D, Nikolouzakis TK, Docea AO, Calina D, Vinceti M, et al. COVID-19, an opportunity to reevaluate the correlation between long-term effects of anthropogenic pollutants on viral epidemic/pandemic events and prevalence. Food Chem Toxicol. 2020;141:111418. https://doi.org/10.1016/j.fct.2020.111418.
Zahra A, Sisu C, Silva E, De Aguiar Greca S-C, Randeva HS, Chatha K, et al. Is There a Link between Bisphenol A (BPA), a Key Endocrine Disruptor, and the Risk for SARS-CoV-2 Infection and Severe COVID-19? J Clin Med. 2020;9(10):3296.
Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. EDC-2: The endocrine society’s second scientific statement on endocrine-disrupting chemicals. Endocr Rev. 2015;36(6):E1–150. https://doi.org/10.1210/er.2015-1010.
Wu D, Tao X, Chen Z-P, Han J-T, Jia W-J, Zhu N, et al. The environmental endocrine disruptor p-nitrophenol interacts with FKBP51, a positive regulator of androgen receptor and inhibits androgen receptor signaling in human cells. J Hazard Mater. 2016;307:193–201. https://doi.org/10.1016/j.jhazmat.2015.12.045.
Marshall NB, Kerkvliet NI. Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci. 2010;1183:25–37. https://doi.org/10.1111/j.1749-6632.2009.05125.x.
• Prasad Singh N, Nagarkatti M, Nagarkatti P. From suppressor T cells to regulatory T cells: How the journey that began with the discovery of the toxic effects of TCDD led to better understanding of the role of AhR in immunoregulation. Int J Mol Sci. 2020;21(21):7849. This review details the current understanding of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunoregulation, including through epigenetics.
Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PloS one. 2011;6(8):e23522.
Schjenken JE, Green ES, Overduin TS, Mah CY, Russell DL, Robertson SA. Endocrine disruptor compounds—A cause of impaired immune tolerance driving inflammatory disorders of pregnancy? Front Endocrinol. 2021;12. https://doi.org/10.3389/fendo.2021.607539.
Murata M, Kang J-H. Bisphenol A (BPA) and cell signaling pathways. Biotechnol Adv. 2018;36(1):311–27. https://doi.org/10.1016/j.biotechadv.2017.12.002.
Araiza V, Mendoza MS, Castro KEN, Cruz SM, Rueda KC, de Leon CTG, et al. Bisphenol A, an endocrine-disruptor compound, that modulates the immune response to infections. Front Biosci (Landmark Ed). 2021;26:346–62.
Csaba G. Effect of endocrine disruptor phytoestrogens on the immune system: Present and future. Acta Microbiol Immunol Hung. 2018;65(1):1–14.
Chalubinski M, Kowalski M. Endocrine disrupters–potential modulators of the immune system and allergic response. Allergy. 2006;61(11):1326–35.
Patrizi B, Siciliani de Cumis M. TCDD toxicity mediated by epigenetic mechanisms. Int J Mol Sci. 2018;19(12):4101.
Tilghman SL, Bratton MR, Segar HC, Martin EC, Rhodes LV, Li M, et al. Endocrine Disruptor Regulation of MicroRNA Expression in Breast Carcinoma Cells. PLoS ONE. 2012;7(3):e32754. https://doi.org/10.1371/journal.pone.0032754.
Curtis SW, Cobb DO, Kilaru V, Terrell ML, Marder ME, Barr DB, et al. Genome-wide DNA methylation differences and polychlorinated biphenyl (PCB) exposure in a US population. Epigenetics. 2021;16(3):338–52. https://doi.org/10.1080/15592294.2020.1795605.
Curtis SW, Cobb DO, Kilaru V, Terrell ML, Kennedy EM, Marder ME, et al. Exposure to polybrominated biphenyl (PBB) associates with genome-wide DNA methylation differences in peripheral blood. Epigenetics. 2019;14(1):52–66.
Yeh C-H, Wu H-C, Kuo T-H, Kuo C-H, Yang S-N, Wang W-L, et al. Suppressive effect on MDC and IP-10 expression in monocytes by endocrine disruptor chemicals. Inflammation. 2010;33(1):10–7.
Skinner MK. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol Cell Endocrinol. 2014;398(1–2):4–12. https://doi.org/10.1016/j.mce.2014.07.019.
Funding
This research was supported in part by grants from the National Institutes of Health (P42ES031007).
Author information
Authors and Affiliations
Contributions
C.M.B. and A.E.E. performed the literature search and drafted the manuscript. R.C.F. critically revised the manuscript.
Corresponding author
Ethics declarations
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Informed Consent
Not applicable.
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environmental Epigenetics
Rights and permissions
About this article

Cite this article
Bulka, C.M., Enggasser, A.E. & Fry, R.C. Epigenetics at the Intersection of COVID-19 Risk and Environmental Chemical Exposures. Curr Envir Health Rpt 9, 477–489 (2022). https://doi.org/10.1007/s40572-022-00353-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-022-00353-9