
Abstract
The mosquito-borne virus Chikungunya has been emerging in new parts of the world in the last decade and has resulted in many new infections and increasing the number of people at risk. Infection with Chikungunya results in Chikungunya fever and possibly the development of arthritis induced by the infection. While the responses of the immune system against Chikungunya infection are not yet fully understood, it has been observed that a CD8+ T cell response is important for viral clearance and protection. Evidence suggests that the arthritis-like symptoms are a result of inflammatory responses of the immune system in the joint tissues where the virus replicates. It is not understood why these symptoms sometimes develop into chronic arthralgia, but it is thought that this may be caused by macrophages infiltrating the joint tissue and resulting in a persistent inflammatory state. Data also implies that inflammation in the joints induces bone erions, which also contribute to arthralgias and arthritis. Since the mechanisms behind the Chikungunya infection-induced arthritis are not yet fully understood, additional efforts should be made to fully comprehend them which also may lead to valuable information on how to eradicate the virus.
Similar content being viewed by others


Introduction
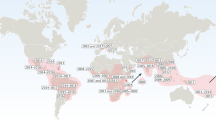
The emerging Chikungunya virus (CHIKV) is a mosquito-borne virus that has infected more than two million people in the last 10 years [1•]. From being a tropical disease restricted to specific geographic areas, it has now become prevalent in previously nonendemic areas, causing greater patient burden. Infection with CHIKV results in Chikungunya fever, and patients develop acute febrile illness, associated with severe polyarthralgias [2]. The word Chikungunya is actually derived from a word in the Kimakonde language which means “to walk bent over” and referred to the people infected with arthralgias who are suffering from severe joint pain [3•].
This review was written to give a brief overview over the timeline of CHIKV outbreaks and the current knowledge on the mechanisms of infection. Additionally, recent findings regarding the T cell responses to CHIKV are also addressed and how the immune responses may possibly contribute to the development of arthralgia.
Emergence of Chikungunya Virus
During the last 60 years numerous CHIKV outbreaks have been documented, although the number of cases might be underestimated since it is thought that the virus was assessed multiple times as dengue virus [4]. The infection was first recognized in East Africa in 1950s [5], and between the 1960s and 1990s, the infection emerged frequently in Central, Southern, and Western Africa [6]. Outbreaks of the virus were also reported in South East Asia between 1960 and 2003. For the first time in 2005–2006, CHIKV emerged in a western country; in the French island La Reunion. It was estimated that 244,000 people were infected and 237 infections resulted in death [7].
In addition, while CHIKV was mainly transmitted by the mosquito Aedes aegypti, it was found that in La Reunion, the virus was primarily transmitted by a different mosquito species, namely Aedes albopictus [8]. Comparing the CHIKV strains from La Reunion and Africa using genome sequencing revealed that a point mutation in the E1 glycoprotein increased the infectivity in Ae. albopictus [8]. Now that CHIKV was able to spread in a new vector, the chance was increased that outbreaks would happen in new areas.
Around the same time CHIKV affected La Reunion, the virus re-emerged in India. It resulted in an outbreak in 13 states with approximately 1.3 million people infected [9]. Although Ae. albopictus was the main vector in islands of the Indian ocean, Ae. aegypti was found to be the main vector in India [10]. Between July and September of 2007, an infected person, traveling from India to Italy, started a series of infections that resulted in the first outbreak in Europe [11, 12]. The virus infected more than 200 persons using the Ae. albopictus vector [13, 14].
In late 2013, the first outbreak of CHIKV was observed in the Americas the caribbean island St. Martin [15]. One year later, more than 776,000 cases have been reported in the Caribbean islands, Latin American countries, and several South American countries [16]. While both vectors Ae. aegypti and Ae. albopictus are prevalent in the Americas, and the virus has the potential to spread by both vector, the E1 A226 V point mutated strain has not yet been observed [17]. Since Ae. albopictus is present in the Americas, it is plausible that mutated strains will arise and more infections might happen.
Pathogenesis
CHIKV belongs to the family Togaviridae and genus Alphavirus, meaning that it consists of positive single-stranded RNA in an envelope. Some alphaviruses are not pathogenic to humans while others are highly infectious. CHIKV belongs within the Alphavirus group to the Old World viruses, which share polyarthritis and a rash as symptoms.
After the virus is inoculated into the body by a mosquito, it is capable of replicating in fibroblasts, skeletal muscle satellite cells, and possibly other skin cells [18, 19]. Evidence is also found for monocyte infection [20]. Replication is similar to other alphaviruses; the target cells are entered by endocytosis and within the endosome the E1 glycoprotein interacts with the host’s membrane, ultimately leading into fusion of the host membrane with the viral envelope and release of the viral RNA into the cytoplasm. The RNA interacts and steers the host’s machinery to produce envelope proteins and replicates of the viral RNA. This is all transported to the plasma membrane of the hosts, and new mature virion particles are released by budding [21].
After CHIKV is inoculated in the body, it disseminates through lymph nodes and microvasculature. Its primary replication occurs in the leukocytes causing leukopenia. The liver and spleen are also considered primary viral replication sites [22]. After primary replication, virus disseminates to the bones, muscle, and articular tissue, generating the acute phase of the disease which is a local inflammatory process [22].
The immune system immediately responds to the presence of the viral replication. Fibroblasts and other infected cells recognize the CHIKV ssRNA using Toll-like receptor 8, 7, and 3, as well as by CARDIF, which is considered to be part of a Rig-like receptor. A type I interferon response is mediated, type I IFNs are produced by fibroblasts [23], and quickly after antibodies are produced against the virus, this immune response is sufficient to clear the virus about 4–7 days after infection [21]. T cell-mediated immune responses are measured about 1 week after infection.
In regard to symptomatology, the onset is usually abrupt and the acute stage presents by sudden onset high-grade fever, arthralgias, myalgias, and skin rash. Swollen tender joints and crippling arthritis are common. In the chronic stage, relapses that include sensation of fever, asthenia, exacerbation of arthralgias, and inflammatory polyarthritis are common. Neurological, ocular, and mucocutaneous manifestations have also been described. In mucocutaneous manifestation, the presentation of conjunctivitis and sometimes generalized lymphadenopathy may be present [24]. The typical duration of joint pain resolves in 1–3 weeks. However, 33 % of patients can persist with arthralgia for 4 months, 15 % for 20 months, and as much as 3–5 years for 12 % of patients [24]. Patients with underlying rheumatic or traumatic joint conditions seem to be more vulnerable to suffering arthralgia for a prolonged period of time [24].
The chronic stage is characterized by unpredictable relapses that include sensation of fever, asthenia, and exacerbation of arthralgias and stiffness. Affected patients may manifest inflammatory polyarthritis, severe subacute tenosynovitis/bursitis (consequently nerve tunnel syndromes) in hands, wrists, and exacerbation of pain on movement in previously injured joints. Older individuals and those with underlying rheumatic and traumatic joint disorders seem to be more vulnerable to develop the chronic stage. Rarely, rheumatic manifestations resulting in joint destruction before resolution after 15 years have been reported [24]. Some studies have documented occurrence of rheumatoid arthritis following Chikungunya fever, suggesting that the viral infection may have a role in the initiation or unmasking of rheumatoid arthritis. This relationship merits further study [24].
T Cell Responses to Chikungunya Virus
To date, T cell immunity after Chikungunya virus infection has not been studied extensively; however, key studies that provide insight into this important aspect of understanding the host response to CHIKV have been conducted and are detailed below. In 2008, Muthuman et al. created a DNA vaccine of E1, E2, and capsid proteins based on consensus sequence of West African, Asian, and Eastern, Central, and South African isolates of CHIKV. Vaccination of C57Bl/6 mice resulted in a robust antigen-specific IFNγ response and IgG response, suggesting that CHIKV structural antigens induce both a T cell and B cell response [25]. Subsequently, Thangamani et al. compared the host immune response to CHIKV when infection was delivered to CD-1 mice either by needle injection or by mosquito bite. Cytokine responses were analyzed by RT-PCR, and it was found that mosquito bites bias the immune response to one that is TH2-mediated as elevated expression levels of IL-4 and IL-10 were found whereas there were decreased levels of IFNγ, IL-2, and TLR-3 expression. Conversely, when CHIKV was administered the animals via needle injection, a TH1-biased immune profile was found as expression levels of IFNγ, IL-2, and TLR-3 were increased and those of IL-4 and IL-10 were lower [26].
Wauquier et al. took the study into a human system as they studied a cohort of 69 Gabonese patients that were acutely infected with CHIKV and examined the resulting immune response, including the T cell response. Where D0 is the onset of symptoms, it was determined that the cytokines IL-2rα, IL-13, IFNα2, and IL-4 are sustained through the acute phase of infection. From D0–D2, the cytokine/chemokine profile that was seen was IL-16, IL-17, PDGF-ββ, IFNγ, and IL-17 with IFNγ peaking on D1 and indicates an early inflammatory response which is consistent with their findings that the population of CD8+ T cells is greatest at this time. Interestingly, CD4+ T cells peaked a bit later by D4 though they did express CD95 suggesting that they may be apoptotic. During the time of D4–D7, the cytokines/chemokines that were predominant were IL-6, IL-8, MCP-1, MIF, MIP-1β, SDF-1α, IL-1rα, G-CSF, GM-CSF, VEGF, and IL-12p40. This milieu is indicative of both a pro-inflammatory T cell response as well as the presence of activated macrophages [27]. Hoarau et al. also investigated the nature of the anti-CHIKV T cell immune response in humans by examining that of 48 patients that were infected with CHIKV during the 2005–2006 La Reunion Island outbreak. They compared the responses between those that had recovered from the infection and those that still suffered from chronic infection and focused on the responses to the proteins E2, nsP1, and capsid. In general, their findings highlighted that the IFNγ response was mostly produced by CD8+ T cells and primarily directed to the C-terminal domain of E2 and to a lesser extent the N-terminal domain of nsP1. The capsid protein induced a significantly lower IFNγ response than the other two proteins. Antigen-specific IL-2 responses were also measured and detected but at a lower magnitude than that of IFNγ [28].
Recent studies further dissected the cellular response to CHIKV so as to understand the roles of the populations and subsets during infection using murine models. Knudsen et al., as part of a broad-based study on immunizations with alphavirus replicons, infected C57Bl/6 mice with wild-type CHIKV and characterized the resulting memory CD8+ T cell populations using MHC class I pentamers associated with an E1 peptide. Their analysis showed that the CD8+ T cell population consisted of primarily a Te or Tem phenotype with the minority, 12 %, was of Tcm phenotype. Additionally, approximately half of the CD8+ T cells were CD27+ CD43+ as opposed to 25 % being of the CD27+ CD43− phenotype [29]. CD27+ CD43+ cells are associated with memory cells that have a high proliferative capacity but are relatively short-lived, whereas CD27+ CD43− are thought to be related to long-term viral control and protection via increased cytotoxic function [30]. The experiments performed by Yee Suan Poo et al. were performed with the goal of elucidating the aspects of the immune response that are responsible for controlling CHIKV infection. Utilizing many different strains of mice with different components of their immune system knocked out, they measured viremia in them after infection with CHIKV. Mice lacking B cells, μMT mice, maintained a set point of viremia (mean of viremeia measurements after 10 days post infection) of 1.6 log10 CCID50/mL of blood, indicating B cells are important for total clearance of the virus; however, it also indicated B cells alone are not responsible for viral control as the viremia in these mice did decrease significantly from their peak values. Rag1−/− mice, those deficient in B and T cells, had a set point viremia of 4 log10 CCID50/mL of blood supporting the notion that T cells are crucial in the control and clearance of the virus. Additionally, NK cells did not appear to have a role in CHIKV clearance in this system [31].
Taking together the conclusions of the aforementioned studies lend to, in general, the following cellular immune response events after infection with CHIKV. CHIKV infection induces a robust T cell response which is primarily CD8+ driven but with a lesser CD4+ component peaking later than the CD8+ and evidence suggests that the T cell response is important for viral clearance and protection. Additionally, the immune profile appeared to be skewed primarily to a TH1-mediated profile dominated by pro-inflammatory cytokines. From the studies that were completed, E1, E2, and nsP1 proteins all seemed to be immunodominant, though studies have not evaluated the cellular response to all proteins after CHIKV infection. Finally, the CHIKV-specific anamnestic response appears to be characterized by mostly an effector and effector memory T cell response.
Overview of Immune Response that Mediates Arthritic Symptomatology
Arthritic pain is the most prevalent symptom that patients manifest during the infection. The recovery from pain and swelling usually takes a few days, but recent studies describe patients with persistence of arthritic symptoms for months or even years [32]. Arthralgia/arthritis usually is symmetric, both in small joints (wrists, fingers) or large joints (knee, shoulder). It can affect multiple articulations simultaneously (polyarthritis) [33]. The virus has tropism to muscle cells producing myalgia in a number of patients [34].
The acute phase is characterized by replication of the virus in target cells which mediate an inflammatory response, which release cytokines and chemokines in the site of the infection causing myositis and arthralgia/arthritis [35]. The secretion of metalloproteinases by these cells in the joint tissue can cause articular damage. Reports on mouse models suggest that those inflammatory cells infiltrate joint, muscle, and similar tissue during the infection, indicating that the muscular and articular damage is mediated by immunopathological inflammatory disorder [36].
Chronic arthralgia and arthritis can have a wide variety of manifestations from minimal restriction of movements to a more drastic persistent incapacitation [37]. Chronic myalgia, arthralgia, and arthritis can occur in 25–55 % of patients. The exact cause of persistence of symptoms in some patients is unknown, but it seems to be associated with the intensity of inflammatory process, extension of articular damage, presence of viral products in joints, and an autoimmune process mediated by target cells which release IL-6 and GM-CSF [38]. Patients with 18 months post illness onset of persistent arthralgia were reported to have long-term infection of perivascular synovial macrophages with Chikungunya virus and infiltration of CD14+ as well [39].
Recently, evidence has shown that a cell-mediated immune response is the primary contributor to persistence of joint manifestations. Infiltrating macrophages in particular have been described to determine the severity and persistence of joint manifestations.
Role of Target Cells in Pathogenesis of Arthralgia
Experimental models of CHIKV-induced arthritis suggest that the pathogenesis is mediated by direct cellular and tissue damage by the virus replication and indirect immune response activation in target tissues. The virus can target joint tissues, osteocytes, myocytes, and chondrocytes. An example of its effects on myocytes is muscle necrosis accompanied by a massive infiltration of inflammatory cells which have been described in Chikungunya mouse models [40].
The macrophage has been described as the main cellular infiltrate in injured tissues [41]. Soluble factors secreted from the macrophage can amplify the inflammatory process recruiting and activating lymphocytes, NK cells to target tissue [41]. Macrophages seem to be the best reservoir in affected tissues, playing a central role in viral-induced arthritis [22, 27].
Immune Response and Inflammatory Mediators to Alphavirus-Induced Pathology
Several mouse studies were done to reproduce the immune response of Chikungunya. During these studies, severe inflammation was noted in the joint, muscles, and bone tissue. This inflammatory process was not altered in mice recombinase-activating gene deficient which lack T and B lymphocytes which shows that the adaptive immunity has a restricted role in the disease pathology of Chikungunya. On the contrary, pharmacological depletion of macrophages in mice caused an annulation of disease symptomatology, causing a lower expression of cytokines (IFN-γ, TNF-α, MCP-1, IL-β, MIP-1α) in the muscle and joint tissue. This demonstrates the greater role of the innate immunity over adaptive immunity in disease progression [36].
Chikungunya-infected mice and nonhuman primates portrayed a release of pro-inflammatory mediators such as IL-6, IFN-γ, TNF-α, and IFN-α/β. During the viremia phase, increase in MCP-1, RANTES, and IP-10 was observed [42]. Additionally, primary human osteoblasts have shown to be susceptible to viral infection causing an induced release of IL-6 and RANKL, while osteoprotegerin was gradually inhibited. The infection of osteoblast by Chikungunya and IL-6 production may contribute to bone loss and the occurrence of arthralgia and arthritis [43].
Several case reports have shown that in few cases, focal bone erosion is caused by Chikungunya virus. In a recent preclinical study in a mouse model of another alphavirus, Ross river virus, there was evidence of periarticular bone loss within the tibial epiphysis and systemic loss in the vertebrae. This was correlated with increased RANKL:OPG ratio and increased osteoclastogenesis [44••]. Recent data suggest that the virus can replicate in the joint tissue and stimulate an immune response inside the bone microenvironment. The synoviocyte-infected cells can drive monocyte/macrophage migration and osteoclast formation [45] causing destruction of the structure [46••]. The osteoblast is very susceptible to the virus, and when infected, it can induce RANKL expression providing the mechanism by which the alphavirus mediates bone loss [43].
In a clinical study with Chikungunya-infected patients from Singapore, the plasma levels of several cytokines (IFN-α, IL-6, IL-12, GM-CSF, IP-10, and MCP-1) correlate with viral load and plasma levels of IL-6 and GM-CSF. Patients with higher levels of cytokines and viral load had persistent arthralgia [47]. A similar study reported that IL-1β and IL-6 are associated with generation of joint pain. The cytokines MCP-1, MIP-1α, and MIP-1β were increased during chronic phase [48].
In Chikungunya-infected patients, the levels of MCP-1, IL-6, and IL-8 were higher in synovial fluid than in sera suggesting that an active monocyte/macrophage trafficking into synovial tissue. Also, high level of matrix metalloproteinase was found in synovial tissue of chronic patients, one of the factors involved in tissue lesion [49].
Complement activation was detected in synovial tissue of infected patients. Levels of C3a marker were higher in infected patients than in patients with noninflammatory osteoarthritis. Recent finding in mouse model of Ross river virus (alphavirus) showed that complement is important to promote inflammatory tissue destruction [40]. Mice which were C3-deficient developed a less severe disease and also had lower levels of skeletal muscle destruction compared to wild-type mice [40]. The C3-deficient mice had a reduced expression of pro-inflammatory proteins S100A9, S100A8, and IL-6. The levels of heterodimeric complex formed by S100A9 and S100A8 were elevated in sera of patients with RA or inflammatory muscle disease in which the expression of these by macrophages was associated with muscle degeneration [50]. Also, in Ross river virus infection, observation suggests that the virus causes complement activation through MBL (Mannose Binding Lectin) pathway contributing to severity of symptoms. Those infected patients with higher MBL levels both in serum and synovial fluid correlated to polyarthritis severity [51].
Type I IFN (interferon) immune response signaling is essential for the control of viral replication and could be an important element to prevent the dissemination of the virus to target tissue causing subsequently arthritis [52]. Mice deficient of type 1 IFN were more susceptible to the infection and had broader viral dissemination [18]. Viperin production (highly inducible endoplasmic reticulum protein that has antiviral activity) from interferon-stimulated gene has shown to be critical in host antiviral response against Chikungunya. Those with deficient Viperin had enhanced viral load and severe joint inflammation.
Conclusion
Chikungunya continues to emerge throughout the globe and is a cause of significant morbidity in rapidly increasing frequency. One of the more debilitating clinical manifestations is arthralgia long after the infection is cleared. Evidence supports a role of the cellular immune response for at least part of the cause of the prolonged joint symptoms. Clearly, the careful development of interventions, such as vaccines, is of utmost importance in combating this disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major Importance
Powers AM. Risks to the Americas associated with the continued expansion of chikungunya virus. J Gen Virol. 2015;96(Pt 1):1–5. Highlights the remergence and spread of chikungunya and its increasing threat.
Staples JE, Breiman RF, Powers AM. Chikungunya fever: an epidemiological review of a re-emerging infectious disease. Clin Infect Dis. 2009;49(6):942–8.
Morens DM, Fauci AS. Chikungunya at the door—deja vu all over again? N Engl J Med. 2014;371(10):885–7. Highlights the remergence and spread of chikungunya and its increasing threat.
Carey DE. Chikungunya and dengue: a case of mistaken identity? J Hist Med Allied Sci. 1971;26(3):243–62.
Robinson MC. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952-53. I. Clinical features. Trans R Soc Trop Med Hyg. 1955;49(1):28–32.
Powers AM, Logue CH. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J Gen Virol. 2007;88(Pt 9):2363–77.
Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3(7):e263.
Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M et al. Two chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS One. 2007;2(11):e1168.
Arankalle VA, Shrivastava S, Cherian S, Gunjikar RS, Walimbe AM, Jadhav SM et al. Genetic divergence of Chikungunya viruses in India (1963-2006) with special reference to the 2005-2006 explosive epidemic. J Gen Virol. 2007;88(Pt 7):1967–76.
Yergolkar PN, Tandale BV, Arankalle VA, Sathe PS, Sudeep AB, Gandhe SS et al. Chikungunya outbreaks caused by African genotype, India. Emerg Infect Dis. 2006;12(10):1580–3.
Angelini R, Finarelli AC, Angelini P, Po C, Petropulacos K, Macini P et al. An outbreak of chikungunya fever in the province of Ravenna, Italy. Euro Surveill. 2007;12(9):E070906.1.
Angelini R, Finarelli AC, Angelini P, Po C, Petropulacos K, Silvi G et al. Chikungunya in north-eastern Italy: a summing up of the outbreak. Euro Surveill. 2007;12(11):E071122.2.
Angelini P, Macini P, Finarelli AC, Pol C, Venturelli C, Bellini R et al. Chikungunya epidemic outbreak in Emilia-Romagna (Italy) during summer 2007. Parassitologia. 2008;50(1-2):97–8.
Watson R. Europe witnesses first local transmission of chikungunya fever in Italy. BMJ. 2007;335(7619):532–3.
CDC.gov 2015: Centers for Disease Control and Pevention. Chikungunya Virus. 2015. http://www.cdc.gov/Chikungunya/index.html.
WHO 2015: World Health Organization. Chikungunya. 2015. http://www.who.int/mediacentre/factsheets/fs327/en/.
Higgs S, Vanlandingham D. Chikungunya virus and its mosquito vectors. Vector Borne Zoonotic Dis. 2015;15(4):231–40.
Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4(2):e29.
Ozden S, Huerre M, Riviere JP, Coffey LL, Afonso PV, Mouly V et al. Human muscle satellite cells as targets of Chikungunya virus infection. PLoS One. 2007;2(6):e527.
Her Z, Malleret B, Chan M, Ong EK, Wong SC, Kwek DJ et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J Immunol. 2010;184(10):5903–13.
Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol. 2010;8(7):491–500.
Assuncao-Miranda I, Cruz-Oliveira C, Da Poian AT. Molecular mechanisms involved in the pathogenesis of alphavirus-induced arthritis. Biomed Res Int. 2013;2013:973516.
Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207(2):429–42.
Mohan A, Kiran DH, Manohar IC, Kumar DP. Epidemiology, clinical manifestations, and diagnosis of Chikungunya fever: lessons learned from the re-emerging epidemic. Indian J Dermatol. 2010;55(1):54–63.
Muthumani K, Lankaraman KM, Laddy DJ, Sundaram SG, Chung CW, Sako E et al. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine. 2008;26(40):5128–34.
Thangamani S, Higgs S, Ziegler S, Vanlandingham D, Tesh R, Wikel S. Host immune response to mosquito-transmitted chikungunya virus differs from that elicited by needle inoculated virus. PLoS One. 2010;5(8):e12137.
Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis. 2011;204(1):115–23.
Hoarau JJ, Gay F, Pellé O, Samri A, Jaffar-Bandjee MC, Gasque P et al. Identical strength of the T cell responses against E2, nsP1 and capsid CHIKV proteins in recovered and chronic patients after the epidemics of 2005-2006 in La Reunion Island. PLoS One. 2013;8(12):e84695.
Knudsen ML, Ljungberg K, Kakoulidou M, Kostic L, Hallengärd D, García-Arriaza J et al. Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J Virol. 2014;88(21):12438–51.
Mikkelsen M, Holst PJ, Bukh J, Thomsen AR, Christensen JP. Enhanced and sustained CD8+ T cell responses with an adenoviral vector-based hepatitis C virus vaccine encoding NS3 linked to the MHC class II chaperone protein invariant chain. J Immunol. 2011;186(4):2355–64.
Poo YS, Rudd PA, Gardner J, Wilson JA, Larcher T, Colle MA et al. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLOS Negl Trop Dis. 2014;8(12):e3354.
Toivanen A. Alphaviruses: an emerging cause of arthritis? Curr Opin Rheumatol. 2008;20(4):486–90.
Queyriaux B, Simon F, Grandadam M, Michel R, Tolou H, Boutin JP. Clinical burden of chikungunya virus infection. Lancet Infect Dis. 2008;8(1):2–3.
Sane J, Kurkela S, Vapalahti O. Chikungunya, a new global epidemic? Duodecim. 2011;127(5):457–63.
Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM et al. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol. 2011;178(1):32–40.
Lidbury BA, Simeonovic C, Maxwell GE, Marshall ID, Hapel AJ. Macrophage-induced muscle pathology results in morbidity and mortality for Ross River virus-infected mice. J Infect Dis. 2000;181(1):27–34.
Borgherini G, Poubeau P, Jossaume A, Gouix A, Cotte L, Michault A et al. Persistent arthralgia associated with chikungunya virus: a study of 88 adult patients on reunion island. Clin Infect Dis. 2008;47(4):469–75.
Suhrbier A, Jaffar-Bandjee MC, Gasque P. Arthritogenic alphaviruses—an overview. Nat Rev Rheumatol. 2012;8(7):420–9.
Trgovcich J, Aronson JF, Johnston RE. Fatal Sindbis virus infection of neonatal mice in the absence of encephalitis. Virology. 1996;224(1):73–83.
Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J Virol. 2007;81(10):5132–43.
Kumar S, Jaffar-Bandjee MC, Giry C, Connen de Kerillis L, Merits A, Gasque P et al. Mouse macrophage innate immune response to Chikungunya virus infection. Virol J. 2012;9:313.
Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P et al. Chikungunya virus arthritis in adult wild-type mice. J Virol. 2010;84(16):8021–32.
Noret M, Herrero L, Rulli N, Rolph M, Smith PN, Li RW et al. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus-infected human osteoblasts. J Infect Dis. 2012;206(3):455–7: 457–9.
Chen W, Foo SS, Taylor A, Lulla A, Merits A, Hueston L et al. Bindarit, an inhibitor of monocyte chemotactic protein synthesis, protects against bone loss induced by chikungunya virus infection. J Virol. 2015;89(1):581–93. Provides evidence of direct implication of anti-chikungunya virus immune responses and arthritis.
Chen W, Foo SS, Sims NA, Herrero LJ, Walsh NC, Mahalingam S. Arthritogenic alphaviruses: new insights into arthritis and bone pathology. Trends Microbiol. 2015;23(1):35–43.
Phuklia W, Kasisith J, Modhiran N, Rodpai E, Thannagith M, Thongsakulprasert T et al. Osteoclastogenesis induced by CHIKV-infected fibroblast-like synoviocytes: a possible interplay between synoviocytes and monocytes/macrophages in CHIKV-induced arthralgia/arthritis. Virus Res. 2013;177(2):179–88. Provides evidence of direct implication of anti-chikungunya virus immune responses and arthritis.
Chow A , Her Z, Ong EK, Chen JM, Dimatatac F, Kwek DJ et al. Persistent arthralgia induced by Chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J Infect Dis. 2011;203(2):149–57.
Chaaitanya IK, Muruganandam N, Sundaram SG, Kawalekar O, Sugunan AP, Manimunda SP et al. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral Immunol. 2011;24(4):265–71.
Santos LL, Morand EF. Macrophage migration inhibitory factor: a key cytokine in RA, SLE and atherosclerosis. Clin Chim Acta. 2009;399(1-2):1–7.
Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006;8(3):R69.
Gunn BM, Morrison TE, Whitmore AC, Blevins LK, Hueston L, Fraser RJ et al. Mannose binding lectin is required for alphavirus-induced arthritis/myositis. PLoS Pathog. 2012;8(3):e1002586.
Seymour RL, Rossi SL, Bergren NA, Plante KS, Weaver SC. The role of innate versus adaptive immune responses in a mouse model of O’nyong-nyong virus infection. Am J Trop Med Hyg. 2013;88(6):1170–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Leroy Versteeg, Maria Eugenia Carter Febres, and Coreen M. Beaumier declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects as performed by any of the authors.
Additional information
This article is part of the Topical Collection on Viral Tropical Medicine
Leroy Versteeg and Maria Eugenia Carter Febres contributed equally to this work.
Rights and permissions
About this article

Cite this article
Versteeg, L., Febres, M.E.C. & Beaumier, C.M. The Role of Cellular Immune Responses on Chikungunya Virus Infection-Induced Arthritis. Curr Trop Med Rep 3, 60–66 (2016). https://doi.org/10.1007/s40475-016-0074-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40475-016-0074-2