
Abstract
Introduction
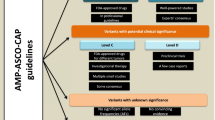
Accurate classification of somatic genetic alterations detected by next-generation sequencing (NGS) assays is of paramount importance to ensure the provision of high-quality clinical data. Clinical significance of variants can be assessed and tiered based on guidelines from the Association for Molecular Pathology (AMP), the American Society of Clinical Oncology, and the College of American Pathology for the interpretation of somatic sequence variants identified in cancer.
Methods
We sought to develop a formal structured approach for the classification of somatic variants in hematologic neoplasms, to account for both a variant’s clinical significance and its ability to drive tumorigenesis, by adapting elements from these existing guidelines. However, we additionally utilized key criteria from the American College of Medical Genetics/AMP standards for variant reporting to focus evaluation into specific categories of evidence and to gauge the effect of a given variant on tumorigenesis.
Results
The combined approach was applied to the annotation of 87 variants identified by a targeted NGS panel for myeloid neoplasms. In the application of our variant evaluation, we classified 2/87 variants as benign, 6/87 as likely benign, 56/87 as variants of unknown significance (VUS), 13/87 variants as likely pathogenic, and 10/87 variants as pathogenic.
Conclusion
Well-established oncogenic alterations were accurately classified as pathogenic. Although there is no defined benchmark for the remaining variants, drawing from two existing guidelines enabled the creation of a modified curation process for variant interpretation that emphasizes systematic review of relevant evidence.

Similar content being viewed by others


References
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–60.
DiStefano MT, Hemphill SE, Cushman BJ, Bowser MJ, Hynes E, Grant AR, et al. Curating clinically relevant transcripts for the interpretation of sequence variants. J Mol Diagn. 2018;20:789–801.
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–24.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Consortium 1000 Genomes Project, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol. 2016;17:9.
Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63.
Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174–83.
Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017;9:4.
Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet. 2016;98:1077–81.
Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genom. 2013;14(Suppl 3):S7.
Kerr ID, Cox HC, Moyes K, Evans B, Burdett BC, van Kan A, et al. Assessment of in silico protein sequence analysis in the clinical classification of variants in cancer risk genes. J Community Genet. 2017;8:87–95.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Houdayer C, Caux-Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat. 2012;33:1228–38.
Tang R, Prosser DO, Love DR. Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Adv Bioinform. 2016;2016:5614058.
Hellen B. Splice site tools: a comparative analysis report. National Genetics Reference Laboratory | Manchester. 2009. http://www.ngrl.org.uk/Manchester/sites/default/files/publications/Informatics/NGRL_Splice_Site_Tools_Analysis_2009.pdf. Accessed 8 June 2021.
Kucukkal TG, Yang Y, Chapman SC, Cao W, Alexov E. Computational and experimental approaches to reveal the effects of single nucleotide polymorphisms with respect to disease diagnostics. Int J Mol Sci. 2014;15:9670–717.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest/Competing interest
Nikita Mehta, Rong He, and David Viswanatha are or were employees of the Mayo Clinic (Department of Laboratory Medicine and Pathology, Molecular Hematology Laboratory) and have no conflicts of interest that are directly relevant to the content of this article. NM is currently employed at Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Ethics approval
The Mayo Clinic Institutional Review Board (IRB) acknowledged that based on responses submitted to the Mayo Clinic IRB eSystem Human Subjects Research Wizard tool, and in accordance with the Code of Federal Regulations, 45 CFR 46.102, this activity did not require IRB review.
Consent to participate
Not applicable.
Consent for publication
The authors all read and approved the manuscript.
Availability of data and material
All variants evaluated in this study are provided in the ESM.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mehta, N., He, R. & Viswanatha, D.S. Internal Standardization of the Interpretation and Reporting of Sequence Variants in Hematologic Neoplasms. Mol Diagn Ther 25, 517–526 (2021). https://doi.org/10.1007/s40291-021-00540-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-021-00540-8