
Abstract
Background
An extensive backlog of pending regulatory decisions is one of the major historical challenges that the South African Health Products Regulatory Authority (SAHPRA) inherited from the Medicine Control Council (MCC). Revising and implementing new regulatory pathways is one of the strategic mechanisms that SAHPRA employs to circumvent this problem.
Objectives
To alleviate the backlog, the use of a new review pathway termed the risk-based review on the scientific quality and bioequivalence assessments was explored. The objective of the study was to articulate the risk-based assessment (RBA) pathway, to determine robust criteria for the classification of the levels of risk for medicines, and to define the improved process to be followed in the assessment and approval of medicines.
Methods
In 2015, an extensive exercise was conducted by SAHPRA to identify the unknown status of in-process applications. The RBA pilot project commenced in 2016 and further piloted in 2021 using the knowledge gained from the 2016 study for optimisation of efficiency.
Results
By 2015 the backlog was quantified as 7902 applications in the pre-registration phase. The 2015 project entailed two phases. The initial phase was conducted to identify the status of 3505 in-process applications, which resulted in the registration of 198 applications. The second phase commenced in 2016 on 4397 applications not yet reviewed whereby the RBA approach was explored. With the developed criteria for risk classification and refined end-to-end registration process, the pilot resulted in a finalisation time with a median value of 90 calendar days and a median approval time of 109 calendar days. The throughput of the RBA pilot study conducted in 2021 was 68 calendar days finalisation time for the 63 applications used. These finalisation times are lower in comparison to the 501 calendar days for the current process employed by SAHPRA for the backlog clearance programme initiated in 2019. Both the 2016 and 2021 studies had similar approval times calculated from the date of allocation of scientific assessments. The reported evaluation timelines for both studies were within 6–7 h for a low-risk quality assessment, 9–10 h for a high-risk quality assessment, 7–8 h for a bioequivalence assessment, and 2–3 h for a biowaiver and initial response assessment.
Conclusions
The refined processes used in the risk-based pilot studies to alleviate the SAHPRA backlog are described in detail. The process managed a reduction of the finalisation time to 68 calendar days in comparison to 501 calendar days for the current process that was employed by SAHPRA for the backlog clearance programme initiated in 2019. The RBA approach, therefore, reduces the finalisation and approval times for quality and bioequivalence assessments for regulatory authorities without compromising on the quality, safety and efficacy of the medicinal products. In addition, the approach provides a prototype solution to counteract the influx of medicinal product applications received by the regulatory authorities.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The South African Health Products Regulatory Authority (SAHPRA) had accumulated a backlog of 7902 medicinal product applications in the system in 2016, and by 2018, this had escalated to 8220. In addition, a median approval time of 1622 calendar days was reported between 2015 and 2018. The growing application backlog in SAHPRA demonstrates the need for drastic interventions; hence the development of the risk-based assessment approach aimed at alleviating the current and continuously forming backlog by reducing overall approval timelines. |
The risk-based assessment approach is a robust end-to-end registration process, which would be a new alternative regulatory review pathway that has been developed to alleviate the backlog and reduce overall approval times. This process includes a risk classification applied before assessments, improved overall registration process, improved evaluation tools, and amended peer-review process. The pilot studies conducted using this new regulatory review pathway confirmed the reduced approval timelines. |
1 Background
In the effort to protect public health, access to free or affordable essential medicines is one of the main obligations by Governments to fulfill the right to health [1]. The World Health Organization (WHO) has reported that one-third of the world’s population does not have timely access to such medicines and has encouraged countries to amend their national legislation or constitutions to provide for this right [2]. Regulatory authorities are established by Governments with a mandate to safeguard the patients by ensuring that safe, efficacious, and quality medicine is accessible at an accelerated rate [2]. The median approval times by several regulatory authorities are outlined in Table 1 for the period of 2015–2019 [3,4,5,6]. The table illustrates the median approval times reported with the lowest as 247 calendar days for 48 applications by the US Food and Drug Administration (FDA) [3], and the highest with a median approval time of 1622 calendar days for 121 New Chemical Entity (NCE) applications by the South African Health Products Regulatory Authority (SAHPRA) [6]. In 2020 a study was conducted by SAHPRA and a median approval time of 790 calendar days was reported for 244 generic applications [7]. Table 1, therefore, demonstrates that SAHPRA has significantly longer approval times compared to other Authorities. The large influx of medicines from pharmaceutical companies due to the emerging pharmaceutical market as a result of the increasing disease burden and the growth of the pharmaceutical generic sector amongst others has made access to medicines a challenge to regulatory authorities in low- to middle-income countries [4, 8].
Regulatory authorities in developing countries such as SAHPRA face a number of resource constraints, with the main one being insufficiently skilled individuals for dossier assessments and manufacturing site inspections. The delays were also attributed to deficient operational processes and increased volume of applications for registration. The long regulatory decision timeframes have serious public consequences, as these delay access to life-saving medicines. In addition, the Medicines and Related Substances Act, 1965 (Act 101 of 1965), Section 22F [9], did not prevent or state how many generics the regulatory authority should register per active pharmaceutical ingredient (API). This Act encouraged ‘dossier farming’ within the industry, which created a significant backlog within the Regulator [10, 11]. SAHPRA received an average of 1200 applications annually between 2006 and 2015 and the authority could therefore not evaluate all the applications received within the period due to resource constraints and other factors as mentioned above. This resulted in the formation of a backlog of applications, delaying access to medicines for patients.
1.1 South African Health Products Regulatory Authority (SAHPRA)’s Organisational Structure
SAHPRA, with internationally recognised standing, is aimed at facilitating the availability, evaluation and approval of the quality, safety and efficacy of medicinal products and related substances intended for humans and animals. In the years in which SAHPRA (formerly Medicine Control Council, MCC) has been in effect, over 20,000 medicinal products have been registered [12]. SAHPRA assumed the roles of both the MCC as well as the Directorate of Radiation Control (DRC) which were housed at the South African National Department of Health (NDoH) [13]. Subsequently, SAHPRA was constituted as an independent entity that reports to the National Minister of Health through its Board [13]. The organisation is headed by the Chief Executive Officer (CEO) with support from the Chief Financial Officer (CFO), Chief Operating Officer (COO), Chief Regulatory Officer (CRO), and the Human Resource Executive, who all form part of the Executive Committee of the organisation (see Supplementary Online Material (OSM) Resource 1). Within the office of the CRO lies the programmes: Pharmaceutical Evaluation Management (PEM), Clinical Evaluation Management, Inspectorate and Regulatory Compliance, and Medical device and Radiation control as illustrated in the OSM (see OSM Resource 2).
The programmes are in turn subdivided into units responsible for coordination and execution of various activities. Within the PEM programme lies the Pharmaceutical and Analytical (P&A) Pre-Registration Unit. The work of the Unit involves the evaluation of the quality and efficacy (bioequivalence) aspects of products submitted as a dossier in the Common Technical Document (CTD) format by pharmaceutical companies. The clinical aspects, i.e., to confirm that the labelling of the generic products is in accordance with the registered innovator products and efficacy of the NCEs is evaluated by the clinical evaluations’ pre-registration unit. Inspection of manufacturing sites is conducted by the Inspectorate Unit. Appropriate naming and scheduling status of the products is conducted by the Names and Scheduling Unit (OSM Resource 2) [14]. The PEM, P&A Pre-Registration Unit has proven to be the rate-limiting part of the registration process since the bulk of the evaluations that include quality and bioequivalence assessments are conducted in the unit. The growing application backlog in SAHPRA demonstrates the need for mechanistic interventions such as the RBA approach to alleviate the backlog by reducing the scientific evaluation timelines.
1.2 Risk-Based Assessments
Risk is defined as the combination of the probability of occurrence of harm and the severity of that harm [15, 16]. The evaluation of risk requires the identification of a hazard and the likelihood of its occurrence [17, 18]. In pharmaceuticals, managing risk is of prime importance to ensure that the patient gets medicines/products of acceptable safety, efficacy and quality, according to WHO standards, as set out in WHO guidelines [15, 16, 18]. Risk assessment is applied on the diseases to be treated as well as in the technology involved in the development and manufacture of the pharmaceuticals. The technology level affects the feasibility of the manufacturing process, including packaging and quality control testing, the overall quality assurance system of the manufacturer, as well as the capacity of the local National Regulatory Authority (NRA) to effectively assess the resultant dossier [19]. Thus, one of the main factors that affect the quality of the product is the quality of the manufacturing process that produces both the API and the Final Pharmaceutical Product (FPP). Hence, sound and reliable processes produce quality products. Quality cannot be tested into the product, but it is to be built into the product during its manufacturing.
In order to expeditiously provide the public with access to quality, safe and efficacious medicines, a risk-based approach to the assessment of a pharmaceutical product should be explored. This approach is discussed in the publication by the Centre for Innovation in Regulatory Science (CIRS), which describes measures that regulatory authorities should consider to apply in the risk-based approach [20]. The review highlights the importance of the level of experience of the evaluators used and the assessment tools employed during assessments to ensure that there is no compromise in the quality and that all critical components are appropriately detailed in the assessments. The component of the level of experience of the evaluators used in the assessments of the dossiers is supported by the results of the project previously undertaken by SAHPRA. In July 2009–September 2010, the Regulator had a backlog of 2114 applications and initiated a project aimed at alleviating the backlog of applications. Only 16.6% of the products were registered while 1.6% were rejected and 6% were cancelled or withdrawn [21]. The reason for the unsatisfactory results were due to substandard reports that were submitted by inexperienced evaluators, which required re-assessment by the PEM, P&A Pre-Registration Unit. This, therefore, illustrates the importance of experienced evaluators who are well knowledgeable with vast experience in the field of regulatory science and scientific assessments with a thorough scientific understanding of the benefit and risk involved [22].
The second component mentioned in the CIRS article is the scientific review tools, which play a major role in the efficiency and effectiveness of the authority and could result in delayed registration, depending on the tools and strategies used to conduct scientific assessments [20]. In the effort to attain shorter registration turnaround times, authorities need to incorporate the benefit-risk factors at the assessment stage. This entails adopting and implementing a systematic process of assessment of the dossier that builds quality into the assessment. Understanding what critical information is needed to reach an acceptable level of certainty to resolve scientific questions and meet regulatory standards for registration is important [22]. Therefore, identification of critical aspects in the Common Technical Document (CTD) and International Conference for Harmonisation (ICH) E3 bioequivalence structures is paramount.
Risk-based assessments, involving the thorough evaluation and reporting of only critical sections in the dossier that affect the quality of the specific product, are now commonly applied by a number of regulators [23, 24]. By applying a risk-based assessment, the following are questions to be considered:
-
What is the risk to the user and how serious is it?
-
What is the weight of evidence that supports that a risk exists?
-
What is the expected and the actual benefit for a specific patient?
-
Will the risk intensify over time?
-
Does the risk outweigh the benefit? [25]
Both practical and theoretical knowledge of regulatory assessment is desirable to achieve a good understanding of the issues likely to be associated with the product under review and identify the risk and the critical aspects [16, 17, 26].
1.3 Objectives
The objectives of the study were fourfold:
-
quantification of the backlog that developed within SAHPRA,
-
defining risk and developing robust criteria for risk classification of products,
-
developing a new robust mechanistic review pathway called the risk-based approach and evaluating the review process based on the results of the pilot study conducted,
-
providing a detailed description of the implementation of the RBA process aimed at reducing the scientific evaluation timeframes and thereby reduce the overall registration turnaround time within SAHPRA.
2 Methods
2.1 The 2015 Backlog Project
The backlog project undertaken in 2015 was divided into two phases. The initial phase entailed the identification of the status of in-process applications and the second phase was on applications not yet allocated for review. The extensive planning of the backlog project required the collaboration of all units involved in the registration process, which resulted in the formation of a backlog working group. The status of most of these applications by the different units was unknown and required an extensive investigation in order to obtain the exact status of the products. The list was created, and the documents were titled in the backlog spreadsheet (Microsoft Excel® 2016, Windows 10), which consisted of all the in-process applications in the pre-registration phase.
2.1.1 Obtaining the Status of In-Process Applications
SAHPRA initiated an overtime project during weekends to allow for the extraction of the information from the registry files, brown files, dossiers, Committee meeting minutes, applicants, etc. For instance, if the product status is unknown, obtaining the information involved the following sequential order, and if it is not obtained in one document area, it moves to the next:
-
the brown files, which should consist of the communications sent to the applicant;
-
the Committee meeting minute documents, which consist of the history and dates of each application discussed and the outcome thereof;
-
registry files, which contain the full history of documents received from applicants were checked to see the available history;
-
if no information is obtained from the above, the applicant was contacted for a re-submission.
It was discovered from this process that a number of units were not aligned when it comes to evaluations, i.e., one unit would have finalised an application while another unit was only at the initial evaluation stage. Therefore, although there might be finalisation in one unit, registration cannot be executed because another unit has not finalised the application. When documentation was obtained from the above four areas, it was promptly shared or communicated with the applicant to facilitate review and accelerated the registration process.
2.2 New Applications—Risk-Based Review
The pilot project was initiated with the available new applications on a first-come first-served basis. During this time, the Authority was allocating applications received in 2011, while those received prior were either registered or in the pre-registration phase under review. There were 208 line-item applications, which equate to 150 master applications that were received towards the end of 2011–2012 that were not yet reviewed. These were used in the pilot study as they were next in the queue to ensure fairness to all applicants. The intent of the pilot study was to observe the effects of the proposed process with the aim of implementing it to all applications upon assessing the results. There were two separate phases within the project, the first one for the in-process applications that was initiated in 2015, and the second phase for the new applications initiated in 2016. For the 2021 pilot study, the applications that were next in line for allocation were in re-submission window eight (8), and were therefore used for further optimisation and efficiency of the process.
3 Results
3.1 The 2015 Backlog Project
For quantification of the backlog, Figs. 1 and 2 illustrate how the backlog resulted within SAHPRA in the period 2006–2015. For example, in 2010, SAHPRA received 1204 applications and could only register 425, resulting in 779 backlog applications. The collective backlog by May 2016 was 7902 applications and only 3779 were registered between 2006 and 2015 [27]. There were 3505 in-process applications in the initial phase for identification of their status and 4397 applications not yet allocated for review in the second phase [27]. The results from these two phases were investigated and the outcomes are detailed below.

A depiction of the registered products within South African Health Products Regulatory Authority (SAHPRA) between 2006 and 2010 resulting in the backlog

A depiction of the registered products within South African Health Products Regulatory Authority (SAHPRA) between 2011 and 2015 further exacerbating the backlog
The backlog pilot project on the in-process applications succeeded in the registration of 198 products, while 189 products were withdrawn by applicants after analysis of the business need. For the 2015/2016 cycle, in quarter one (April–June 2015), 34 products were registered, in quarter two (July–September 2015), 43 products were registered, in quarter three (October–December 2015), 88 products were registered, and in quarter four (January–March 2016), 33 products were registered. The project achieved the clearance of 387 products in 2015 as well as obtaining the status of all the applications that were pending registration (see Fig. 3). The 448 registered applications include 250 registrations via the normal process that were not part of the pilot project.

Status classification and quantification of the in-process applications once Phase 1 of 2015 project was concluded. GMP Good Manufacturing Practice, P&A Pharmaceutical and Analytical pre-registration Unit
Figure 3 shows the grouping of the status of applications obtained during the 2015 project. The exercise managed to identify and classify the status of all pending applications, a task that was historically difficult for the authority. The authority did not have a central database or tracker for applications and relied on individual units to monitor the applications, which led to misalignment within the units as they were not communicating with one another on evaluations of applications. As a result, there were 707 applications with P&A finalised status, and 519 applications with Clinical finalised status. There were also 244 applications with P&A and Clinical finalised status; however, these could not be approved since the Inspectorate and Names and Scheduling Units had not finalised the applications. These applications were classified as ‘the low hanging fruits’ since they were near registration and only required finalisation by one or two units. For the P&A finalised applications, it meant that other units needed to focus on those products to attain registration and vice versa for the other finalised groups.
3.2 Risk-Based Assessment Process
3.2.1 Registration Process
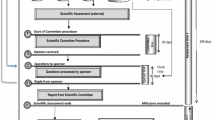
Once the status of the pending applications was concluded, the authority moved on to reviewing the evaluation pathways for the new applications. Strategic planning over a 2-year period between 2014 and 2016 was employed in order to alleviate the backlog by improving the existing registration process. It was important that the process be revisited to ensure that the proposed process is seamless and avoids the formation of a backlog in future. The overall developed and refined process as detailed in Fig. 4 involved changes to the previous practices, thereby promoting efficiency and timely access of medicines to patients.

Proposed risk-based assessment end-to-end registration process in the Pharmaceutical Evaluation Management, Pharmaceutical and Analytical Pre-Registration Unit for quality and bioequivalence assessments. The process is repeated for the response cycle and only 10 working days are allocated for the second response cycle. HPA Health Products Authorisation, PC Portfolio Coordinator
3.2.2 Risk Classification
Upon re-assessment and refining of the two pilot studies for scale-up and implementation in the BAU section of SAHPRA, the risk classification template was refined through consultation with numerous experts and extensive literature review [19, 28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. This resulted in the developed risk classification template (Table 2) used for determining the risk of generic products including essential medicines that qualify to be included in this pathway. The model and structure detailed in the concept paper by the WHO was used, in which a scoring is assigned for each aspect to consider and the overall scores was used to determine the risk class of the product using Table 2 [19]. Table 3 indicates the risk classification matrix employed to deduce the overall outcome. Note that before the 2021 pilot study, it was decided that NCEs, biologicals medicines or biosimilars will not be reviewed using this pathway; a full review would be conducted for these applications.
For the products that were part of the pilot studies, the overall risk classification of products was deduced using Table 3 and overall classification identified.
From the findings reported, evaluation templates were designed according to the level of risk for evaluators, clearly identifying critical sections for the different risk classifications. The templates are included as OSM Resources 3 and 4. The sections that are critical are identified in the Discussion section.
3.2.3 Summary of Results on the Risk-Based Approach
Table 4 provides a summary of the results from the backlog pilot project conducted in September 2016 and September 2021 by SAHPRA. There were ten evaluators used in both pilot studies; for the 2016 pilot, seven were external evaluators and three were internal evaluators, while for the 2021 pilot study eight were external and two were internal evaluators. The reported finalisations times and approval times for both studies are depicted in Fig. 5, which illustrates the median values for the finalisation times in both pilot studies as well as the reported minimum and maximum times. A number of outliers are witnessed in the depictions for applications that took longer to finalise than the other applications due to applicants not addressing the queries as required. Delays in approval times after finalisations are attributed to other units not yet finalising the products, hence delaying registration. This also illustrates how the rate-limiting PEM, P&A pre-registration unit managed to finalise applications before other units, which has always been a historic problem.

The distribution of finalisation times and approval times for applications in the backlog Phase 1 (2016) and 2021 pilot studies. Box: 25th and 75th percentiles. Whiskers: 5th and 95th percentiles
Table 5 provides the outcomes of the risk classification of the products that were in the two risk-based assessment pilot studies. This shows that the classification largely depends on the dosage form of the product and the manufacturing process of the final product as stated by Tran et al. [32].
3.2.4 Assessment Timelines
The assessment times were recorded for each application. Figure 6 illustrates the median times obtained for assessment of a simplified low-risk application, high-risk application, bioequivalence assessment, biowaiver assessment and a response assessment. For the 2021 pilot study, four of the applications were omitted from the calculations since two were clones of already registered products and two had pre-approvals by the PEM, P&A pre-registration unit before February 2018, and only minor variations were submitted for review. Hence, the total n value was 59, which is 38 low-risk applications and 21 high-risk applications (Fig. 6). It should be noted that a Phase 2 pilot study was conducted in 2022 in order to monitor upscaling of the number of applications to 156; a different template was used and is included as OSM Resource 5. This was pre-populated by the applicant and used as an evaluation template for quality assessments. The reported evaluation times for the second phase in 2022 was a median time of 14 h for high-risk and 10 h for low-risk applications. The BE, biowaiver and response assessments remained the same as the templates in the 2021 pilot study results.

Median evaluation times reported in the two risk-based assessment pilot studies for low-risk, high-risk, bioequivalence (BE), biowaiver and responses. (n) = number of product applications. Box: 25th and 75th percentiles. Whiskers: 5th and 95th percentiles
4 Discussion
4.1 2015 Backlog Project
For the initial phase of the project, the identification of the status of each pending application proved to be a success as it allowed for better coordination and management of applications. In addition, obtaining the status of the finalised products from each unit provided a list of applications that each unit can focus on (Fig. 3). Although allocation was conducted at the same time by the Health Products Authorisation (HPA) section, the units did not initiate the evaluations at the same time. With the improved process this would be alleviated as communication to the applicant will be synchronised for all the applications.
4.2 New Applications—Risk-Based Assessments
The planning of Phase 2 of the 2015 backlog involved engagements with other stakeholders for the success of the project. The stakeholders, such as the applicants and the Expert Committees, held a wealth of knowledge regarding processes, historical information, industry insight, and in the planning and execution of the project for new applications. It was therefore imperative that they were consulted in the decision-making of the project to allow for a seamless process to occur. The proposed process was outlined, and modifications were made where necessary until a consensus was reached to initiate the pilot project.
The proposed process was communicated with all stakeholders involved, which included the CEOs of the pharmaceutical companies in the pilot study, the P&A Expert Committee Members and Unit, the Clinical Evaluations Expert Committee Members and Unit, the members of the MCC Registration Committee, and the Industry Technical Group (ITG). It was agreed that all new applications not yet reviewed should be resubmitted to facilitate review. This is because the submission for these products were between 2011 and 2012, thus the information in the dossiers was outdated. It was observed that the frequent recommendations for the old applications, since 5 years had lapsed, were on updates of the stability data, updated Certificate of Suitability (CEP), changes in the methods of synthesis, changes in the API manufacturers, changes in the FPP manufacturers, etc. This meant that several changes had occurred to a product over time, and in some instances, the product was considered non-existent as the final product manufacturers were no longer in business or were no longer manufacturing it. Thus, after registration, the applicant would apply for post-registration amendments, and by registering the products that essentially no longer exist, MCC was shifting the work to the Post-Registration Unit without eliminating the burden the authority faced. Hence, applicants were requested to uplift, update and re-submit the paper documents. Uplifting of the paper dossiers was conducted 2 months prior to the re-submission date, which gave applicants enough time to update their applications.
Consultation with the applicants resulted in withdrawal of 31% of the applications due to the lack of a business need for the product and only 99 master applications were left for the pilot study. The dossiers were re-submitted between 12 and 16 September 2016, distributed to the respective units, and evaluated by the PEM, P&A Pre-Registration Unit during the evaluation week held on 19–23 September 2016.
Even with the two phases as detailed above, by 2018 the backlog of applications had increased to 8,220. In 2018, the authority embarked on a project called the Backlog Clearance Programme aimed at clearing the existing backlog over a specified time. The planning and development of the project was initiated in February 2018 through the assistance of a project consulting firm, which assisted in the quantification of the backlog. Inherited processes and practices from the former MCC were re-assessed and the backlog project was initiated in August 2019 to support new methodologies required to achieve the goal of clearing the backlog of applications [7]. The project was initiated through the assistance of funding from government, development partners and donors [48].
The applicants were initially requested to indicate if they would like to include their applications in the Backlog Clearance Project. Upon analysis of the business need and proposed timeframe to submit there were 4,610 applications that opted out of the project and 99 applications were withdrawn. Not being part of the backlog project meant once the dossier was ready for resubmission with the new requirements, it would be submitted to the BAU section of SAHPRA. The in-process applications that were near finalisation, by either unit, were assessed in the BAU and concluded. Thus, SAHPRA initiated the Backlog Clearance Project in August 2019 with 3,343 applications, which translates to 1,364 master applications.
The Backlog Clearance Project utilised 56 external domestic and international evaluators to conduct the scientific assessments as well as the internal evaluators from the BAU section working overtime to assist with the project. By May 2021, 34% of the applications had been cleared. This was nearly 2 years after the initiation of the project where the intent was to eliminate the backlog in 2 years. The program was extended by 1 year and 5 months to December 2022 and the delay in the clearance was attributed to the assessments conducted within the PEM, P&A Pre-Registration component due to the bulk of the work being done in this unit [49]. Hence, the necessity for the refinement of the risk-based assessment in September 2021 in an effort to conclude the Backlog Clearance Project in the set time. The 63 applications that were next in line for allocation were in re-submission window eight (8) and were therefore used in the 2021 pilot study.
In 2019 when the backlog clearance programme was initiated, the business-as-usual (BAU) section was provided with the opportunity to start on a clean slate while the backlog clearance programme dealt with all the ~8,220 applications. In the period 2019 to 2022, SAHPRA amended its processes and put systems in place such as the inclusion of a tracker that allows all units to monitor each other; however, even with that, a backlog formed within the BAU section of SAHPRA. The tracker was aimed at providing transparency and synchronisation within the units; however, this did not correct the misalignment as units could still allocate the same applications at different times and communicate the queries at different times. The solution to this would have been to have one set of queries from the different units communicated at the same time by the PC, as conducted in the 2015 study to ensure alignment within units at all times. This meant some units would finalise applications before others, which would lead to misalignment. It should be noted that the root cause of the backlog was not as a result of one factor such as the misalignment of units only, there are a number of reasons, which are detailed in the study, and which is why the risk-based assessment approach was developed as an end-to-end registration process providing corrective or preventative measures or solutions to prevent the root causes from occurring in future.
4.3 Risk-Based Assessment Process
4.3.1 Registration Process
A reassessment of processes was necessary for the authority for improved efficiencies. An improved registration process was employed as detailed in Fig. 4.
The following were improved in the developed process illustrated in Fig. 4:
-
Previously, the units were only allocated an application by the HPA, thereafter communication with the applicants would be made by the separate units. A Portfolio Coordinator (PC) responsible for coordinating and collating outcomes from the units was introduced as one communication to the applicants.
-
The introduction of the Inspectorate Unit confirming the Good Manufacturing Practice (GMP) status before allocation to other units was included since previously this would only occur once the scientific assessments had been concluded by the PEM, P&A and clinical evaluations of Pre-Registration Units. The inspections being conducted towards the end of the process would further delay the registration of applications.
-
The use of a risk-based approach to conduct scientific assessments to reduce the assessment times by the PEM, P&A Pre-Registration Unit with assessments focused on the critical quality attributes of the product.
-
The use of a pre-populated evaluation template to aid in the reduction of evaluation times. This allowed for the technical person to screen the applications to check if the updated information, such as the updated stability data, is as per the requested shelf-life, the updated Certificate of Suitability (CEP) is included, etc.
-
Frequent peer review meetings. For the 2016 pilot study, an evaluation week approach was used where a week was blocked for evaluation, during which towards the end of each day evaluators discussed the reports and query letters sent to the HPA. This promoted scientific knowledge sharing and ensured that queries going out to the applicants were critical aspects to be addressed in the dossier and that the queries were standardised. This was only conducted once, and the rest of the applications awaited the P&A Committee meetings held on a 6-weekly basis. This resulted in some delays.
In the refined process in 2021, weekly peer review meetings were introduced, which allowed for better throughput of query letters to the applicants. The selection of the date for each peer review session was based on the availability of evaluators using the When Available poll. The reports were then compiled into meeting documents and uploaded on Google Docs well in advance to allow evaluators to provide their comments. The living document would then show all comments in real-time, allowing all evaluators to see each other’s comments. This assisted in drastically reducing the meeting sessions as only specific points of discussion, highlighted by the peer-review panel, were discussed. Most other aspects were collaboratively deliberated on during the real-time discussions via Google Docs.
-
The response time was reduced from 90 calendar days to 30 calendar days and only two response cycles were allowed, which the pharmaceutical companies agreed on for the 2016 study.
In the refined process this was further reduced to 10 working days; however, applicants could request an extension if required. The requests for extension were for 41% of the responses, therefore the response timeline was increased to 15 working days for initial responses and 10 working days for further responses.
Once this robust process had been concluded, the products were classified according to risk.
4.3.2 Risk Classification
Ahead of assessing the aspects of the API and FPP, prior work conducted by other NRAs or Regulatory Institutions should be considered. Recognition of the work previously done is termed as reliance. And, according to the WHO, reliance is defined as the act whereby one regulatory authority in one jurisdiction may consider and give significant weight to totally or partially rely upon scientific assessments or inspection reports performed by another authority or trusted institution in reaching its own decision [20]. The relying authority uses this work according to its own scientific knowledge and regulatory procedures and retains its own regulatory responsibilities. Historically, SAHPRA had not implemented this review pathway until 2019 when the backlog clearance programme was initiated [48]. The authorities with which SAHPRA aligns itself and uses the unredacted reports of are the European Medicines Agency (EMA); Health Canada; Medicines and Health Products Regulatory Agency (MHRA) in the United Kingdom; Ministry of Health, Labour and Welfare (MHLW) in Japan; Swiss Agency for Therapeutic Products (Swissmedic); Therapeutic Goods Administration (TGA), Australia; and the FDA [50]. SAHPRA is also currently utilising partial reliance through the use of submissions such as CEPs by the European Directorate for the Quality of Medicines (EDQM) and Certificates of Prequalification (CPQs) of the API by the World Health Organisation Prequalification Team: Medicines (WHO PQTm). The developed template in Table 2 therefore accommodates the reliance aspect as well during risk classification.
The non-reliance critical aspects are also considered during quality and efficacy (bioequivalence) aspects of products submitted for approval, and detailed below to assist in the overall classification of the product.
When it comes to defining the risk pertaining to the API, the following key aspects of the API are assessed:
-
Availability of a valid CEP/CPQ (Certificates of Prequalification (CPQs))
-
Pharmacopoeial status of the API
-
Biopharmaceutics Classification System (BCS) of the API (in particular aqueous solubility)
-
Solid state properties (solubility, hygroscopicity, particle size distribution (PSD) and polymorphism)
-
The concentration of the API in the FPP.
The key aspects to be considered in the FPP are:
-
Pharmacopoeial status of the FPP
-
Type of dosage form
-
Complexity of the manufacturing process
-
Excipients
-
Container closure system (CCS).
The key aspects in the bioequivalence study:
-
The bioequivalence (BE) with the reference products and comparative dissolution with the reference products.
Based on the identified aspects to consider as stated in Table 2, a product could be classified as low or high risk.
4.3.3 Critical Areas to be Reviewed for Low-Risk Products
A combination of literature reported by Tran et al. [32] and the concept paper by the WHO [19], as well as a wide array of expert advice garnered on the approach, categorically assisted in the determination of the critical attributes of manufacturing and overall risk ranking of the product. With this information, the CTD sections and extent of evaluation thereof could be established. The areas of concern have been included below and will be thoroughly evaluated for low-risk applications. The relevant templates are used for assessment with the critical sections included.
The identified critical sections of the CTD for low-risk applications are as follows:
-
Module 1.3 Labelling and packaging (Professional Information (PI), Patient Information Leaflet (PIL) and Label)
-
Quantitative and qualitative composition
-
Storage conditions
-
Container closure system
-
Appearance
-
-
Module 1.7.4.1 Batch Release
-
API and Inactive Pharmaceutical Ingredient (IPI) batch release
-
Release (Final Product Release Control (FPRC)/Final Product Release Responsibility (FPRR))
-
-
Module 1.10 Foreign regulatory status
-
Marketing authorisation information for reliance
-
-
Module 3.2.S. Active Pharmaceutical Ingredient
-
3.2.S.1.3 Physico-chemical properties (depending on dosage form)
-
3.2.S.2.2 Method of synthesis (N/A if CEP/CPQ is submitted)
-
3.2.S.3.2 Impurities (N/A if CEP/CPQ is submitted)
-
3.2.S.4.1/2 Specifications (N/A if CEP/CPQ is submitted, however, assess the API specifications by the FPP manufacturer)
-
3.2.S.7 Stability (N/A if retest period is stipulated on CEP/CPQ)
-
-
Module 3.2.P Finished Pharmaceutical Product
-
3.2.P.1 Components and composition of the final product
-
3.2.P.3.3 Manufacturing process/Batch Manufacturing Record (BMR)
-
3.2.P.5.1 Specifications
-
3.2.P.7 Container closure system
-
3.2.P.8 Stability
-
-
Bioequivalence
The sections proposed for the bioequivalence section are included below and are in line with ICH and EMA requirements [51,52,53]. In the case where a BCS-based biowaiver is requested (BCS class I and III applications), only two sections would be assessed. These include the details of the test and reference product used in the study and comparative dissolution profiles, thus reducing the assessment review times. This template, used as an evaluation tool, would reduce the current reported evaluation timelines, as it is designed to point out and discuss critical aspects of the biostudy.
The identified sections from the bioequivalence template are as follows:
-
Details of the test and reference product used in the study (applicable for biowaiver request)
-
Comparative dissolution profiles (applicable for biowaiver request)
-
Study method and design
-
Summaries of statistical and pharmacokinetic data
-
Bioanalytical report parameters.
Certain sections are excluded from evaluation for low-risk applications. The rationale for these exclusions, which addresses the risk mitigation for each, are as follows:
-
Batch analyses (3.2.S.4.4 and 3.2.P.5.4) are not evaluated for low-risk applications because the stability results (3.2.S.7.3 and 3.2.P.8.3) at the initial time point essentially serve as batch analyses. In addition, the impurities section also includes profiling of the impurities and residual solvents formed, thus these sections mitigate the risk since they are assessed.
-
Reference materials sections (3.2.S.5 and 3.2.P.6) are for documentation purposes and do not need to be assessed since the API would have been confirmed already in preceding sections, such as the method of synthesis, impurity section and specifications. In most cases, 3.2.P.6 refers to section 3.2.S.5 of the dossier. The working standard and primary standards are those manufactured by the applicant and synthesis would, therefore, be in line with the proposed methods.
-
Pharmaceutical development (3.2.P.2) is not assessed for low-risk applications, because this is research and development conducted by the manufacturer for optimisation of the final manufacturing process for commercial product/s. The final proposed manufacturing process is then assessed in section 3.2.P.3.3 and the information is verified by the batch manufacturing records. In addition, for the oral solid dosage forms that require the submission of a bioequivalence study, certain critical aspects of the pharmaceutical development section are evaluated. These include in vitro dissolution studies as these are covered in the bioequivalence template for evaluation. For solid oral dosage forms, selection of inactive pharmaceutical ingredients (IPIs) is covered by the bioequivalence assessment where similarity to the reference product is reviewed, and in the case where the excipients are not similar to the reference product, API-excipient compatibility should be confirmed under 3.2.P.2. In the case of liquid dosage forms, excipient similarity to the reference is confirmed under Module 3.2.R.1.4.1 and in the case where the excipients are not similar to the reference product, API-excipient compatibility would be confirmed under 3.2.P.2. The designed templates therefore provide guidance for these.
-
Module 3.2.P.3.1 details the full name and address of the final product manufacturer. The name of the final product manufacturer is confirmed in the administrative table at the beginning of the pre-populated template. In addition, the Inspectorate Unit confirms and validates this during inspections.
-
Batch formula (3.2.P.3.2) is not assessed since it is confirmed during assessment of the batch manufacturing records, which consist of actual quantities of API/s and IPI/s used for the proposed batch(es).
-
Validation of analytical methods (3.2.S.4.3 and 3.2.P.5.3) is not assessed because the product would be pharmacopoeial and only verification is then required. In addition, specification limits provided found to be within ICH requirements will be confirmed since the specification section is assessed for low-risk applications. At most, the evaluator may only confirm the submission of the reports for noting for low-risk applications.
4.3.4 Critical Areas to be Reviewed for High-Risk Products
If a product is classified as high risk, additional sections over and above the ones identified for low risk would also require thorough evaluation and reporting on the respective templates. The additional sections to assess for high-risk products include the following:
-
Module 1.3 Labelling and packaging (PI, PIL and Label) – same as low-risk
-
Module 1.7 Good Manufacturing Practice – same as low-risk
-
Module 1.10 Foreign regulatory status – same as low-risk
-
Module 3.2.S Active Pharmaceutical Ingredient
-
3.2.S.4.3 Validation of analytical methods for the API – additional section for high-risk applications
-
-
Module 3.2.P Finished Pharmaceutical Product
-
3.2.P.2 Pharmaceutical development of the FPP
-
3.2.P.3.5 Process evaluation of the FPP validation
-
3.2.P.5.3 Validation of analytical methods for the FPP
-
3.2.P.7 Container closure system (for sterile applications)
-
-
Bioequivalence
-
Details of the test and reference product used in the study (applicable for biowaiver request)
-
Comparative dissolution profiles (applicable for biowaiver request)
-
Study method and design
-
Summaries of statistical and pharmacokinetic data
-
Bioanalytical report parameters
-
The justification stated above for the sections that are not to be assessed are also applicable for high-risk applications. Note that risk classification will not be applied to NCEs and biological applications; instead full review will be conducted due to the criticality of the medicines.
4.3.5 Summary of Results on the Risk-Based Approach
In the second phase of the 2015 backlog pilot project for new applications, all 99 master applications were finalised within 9 months, with the median time calculated as 90 calendar days. The outliers were noted as 7, 8 and 9 months as indicated in Fig. 5. These were due to the FPP manufacturers receiving a negative status and therefore inspection had to be arranged by the Inspectorate Unit before evaluation could take place. There were other instances where the applicants requested an extension to submit responses, and this led to the delay in finalisation. For the refinement of the process in 2021, a median finalisation time of 68 calendar days was obtained (Fig. 5). Of the 63 applications, six were withdrawn while in-process in the response phase. However, the initial evaluation was already conducted for these so they were included in the calculations of evaluation times.
From the 63 applications, 21 applications were classified as high risk and 42 classified as low risk as depicted in Table 5. From Table 5, it is observed that all immediate-release tablets and capsules were low risk, which constitute 51% of the applications. From the 90% generic applications that SAHPRA receives, most of these are pharmacopoeial and well-known with readily available extensive research conducted on them; therefore, due to this, classification would be low risk. In addition, the dosage forms were not novel, therefore overall classification was low risk. The same applies for the other dosage forms classified as low risk.
4.3.6 Assessment Timelines
Figure 6 illustrates the reported evaluation times by the evaluators who were part of the two risk-based assessment pilot studies in 2016 and 2021. The graphical depiction shows the calculated median values as 6.3 and 7.0 h in 2016 and 2021, respectively, for low-risk quality assessment timelines. As observed from Table 4, products classified as low risk were immediate-release tablets and capsules, topical gels, mouth wash, throat spray, oral syrups and oral solutions. The median values for high-risk quality assessments were reported as 9.5 and 10 h from the two pilot studies, respectively. Products classified as high risk were sterile intravenous injections and infusions, ophthalmic solutions, delayed-release tablets and sterile lyophilised powders. The bioequivalence study assessment times were 8.4 and 8.0 h using the proposed template and biowaivers reported as 2.3 and 2.6 h with initial response assessment times as 2.6 and 3.4 h. The calculations above were based on a simplified submission that contains one API from one API manufacturer who submitted an Active Pharmaceutical Ingredient Master File (APIMF), with only one FPP manufacturer applied for. In a case where a CEP was submitted, the median evaluation times were 5–6 h for low-risk and 7–8 h for high-risk products; when two APIMFs were submitted, the evaluation times were 11–12 h for low-risk and 13–14 h for high-risk products. This resulted in the deduction that one APIMF assessment takes 4–5 h and one FPP takes 5–6 h to assess for high-risk applications. The reported medians have resulted in a reduction in the assessment times without the compromise to quality as only critical sections that will impact the quality of the product are adequately assessed.
For the Phase 2 pilot study conducted in 2022, the quality assessment timelines for high risk is reported as a median of 14 h and 10 h for low risk. The increased assessment timeline is due to the different quality template used that has been pre-populated by the applicant. The evaluators therefore would spend time validating the information populated by the applicant with the scientific information in the dossier to ensure that accurate information was completed.
Once applications that undergo the risk-based assessment pathway are registered, the following post-marketing surveillance or monitoring procedures were proposed and will be conducted:
-
The applicant will be requested to provide the Post-Registration reports on a yearly basis to Pharmacovigilance and annual product review report to the Inspectorate Unit. Depending on the information submitted on the reports, the Inspectorate could perform inspections of the non-compliant manufacturer/applicant.
-
Ongoing post-marketing surveillance will be conducted on the products by the Inspectorate Unit.
-
Re-evaluation of the information (dossiers) after 5 years will be conducted on all applications.
5 Conclusions
The large influx of applications as a result of ‘dossier farming’ as well as resource constraints experienced by SAHPRA over the years resulted in the formation of a backlog as large as 8220 applications. The organisation needed to implement drastic changes in order to reduce the timelines to promote timely access to medicines. A backlog pilot project was conducted in 2016 to alleviate the existing backlog of applications at the time. The pilot project consisted of 99 master applications and managed to reduce the finalisation timelines to a median value of 90 calendar days. The refined and efficient process was described in detail as well as the knowledge gained from the project. These learnings were used in the refined and optimised risk-based assessment pilot study in 2021. This pilot study was initiated with applications from re-submission window 8 of the Backlog clearance programme project initiated by SAHPRA in 2019. The study was resumed with 63 applications and a median finalisation time of 68 calendar days recorded, which is significantly lower compared to the initial pilot study (90 calendar days) and the current process employed by SAHPRA for the backlog clearance programme initiated in 2019, which resulted in the finalisation time of 501 calendar days. The risk-based approach is discussed in detail as it involves the robust risk classification matrix to employ that allows for the categorisation of a product to the appropriate risk class. The approach also details which sections of the CTD and bioequivalence study are considered critical for comprehensive assessment. The identified sections for the assessment of the two risk classes ensures that quality, safety and efficacy are not compromised while accelerating access to medicine for patients. The risk-based approach therefore essentially aims to reduce the finalisation timelines for quality and bioequivalence assessments for authorities, which will greatly reduce the overall registration timelines. Implementation of this approach by other regulatory authorities will assist in the reduction of the backlog of applications created due to resource constraints and the large influx of applications that are of urgent need for the public.
References
Milani B, Scholten W. The world medicines situation 2011: access to controlled medicines, Department of Essential Medicines and Pharmaceutical Policies, 3rd ed. WHO, Geneva, 2011. Available: https://www.who.int/publications/i/item/WHO-EMP-MIE-2011-2.4. Accessed 25 June 2022.
World Health Organization (WHO), Medium-term strategic plan 2008–2013, Geneva, 2013. Available: http://apps.who.int/gb/ebwha/pdf_files/MTSP-08-13-PPB-10-11/mtsp-3en.pdf. Accessed 25 June 2022.
Roth L. Department of health and human services, FDA Drug Review Timeline Transparency; Statement of Policy, Federal Register, Vol. 86, No. 10, 2021.
Rodier C, Bujar M, McAuslane N, Liberti L, Munro J. Centre for Innovation and regulatory Science; New drug approval for six major authorities 2010-2019: Focus on Facilitated Regulatory Pathways and Internationalisation, R&D briefing 77, version 2, 2020.
Patel P, Cerqueira D, Santos G, Soares R, Sousa V, Liberti L, McAuslane N. A baseline analysis of regulatory review timelines for ANVISA: 2013–2016. Ther Innov Reg Sci. 2020;54:1428–35.
Keyter A, Salek S, Gouws J, Banoo S, Walker S. Evaluation of the performance of the South Africa regulatory agency: recommendations for improved patients’ access to medicines. Ther Innov Reg Sci. 2020;54:878–87.
Keyter A, Salek S, Danks L, et al. South African regulatory authority: the impact of reliance on the review process leading to improved patient access. Front Pharmacol. 2021;12:1–11.
Mikulic M, Pharmaceutical market: Revenue of the worldwide pharmaceutical market from 2001 to 2021, Statista. 2022. Available: https://www.statista.com/statistics/263102/pharmaceutical-market-worldwide-revenue-since2001/#:~:text=As%20of%20end%2D2021%2C%20the,what%20people%20pay%20for%20medication. Accessed 17 September 2022.
South African Health Products Regulatory Authority (SAHPRA): Medicines and Related Substances Act, 1965 (ACT 101 OF 1965), Government Gazette 40869, May 26, 2017. https://www.sahpra.org.za/wp-content/uploads/2020/02/Government_Gazette_Medicines_and_Devices_Act_Jun_2017-1.pdf. Accessed 01 April 2022.
Leng H, Pollock A, Sanders D. The impact of the Medicines Control Council backlog and fast-track review system on access to innovative and new generic and biosimilar medicines of public health importance in South Africa. S Afr Med J. 2016;106:350–3.
Molokwane MF. The effect of dossier farming on medicine registration in South Africa, University of the Western Cape, August 2020. http://etd.uwc.ac.za/xmlui/handle/11394/7872 Accessed 01 April 2022.
South African Health Products Regulatory Authority website, Registered Health Products list: https://www.sahpra.org.za/registered-health-products/ Accessed 01 March 2022.
South African Health Products Regulatory Authority website, Organisational history and structure: https://www.sahpra.org.za/who-we-are/. Accessed 01 March 2022.
South African Health Products Regulatory Authority (SAHPRA): Fit for purpose structure, 2020/2021, 2021. pp1-15.
World Health Organisation (WHO). Annex 6, Good practices of national regulatory authorities in implementing the collaborative registration procedures for medical products. 2014. https://www.who.int/docs/default-source/medicines/norms-and-standards/guidelines/regulatory-standards/trs1019-annex6.pdf?sfvrsn=f839be632. Accessed 02 July 2022.
World Health Organization (WHO). WHO guidelines on quality risk management. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations. Forty-seventh report. Geneva, 2013: Annex 2 (WHO Technical Report Series, No. 981), 2013.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Q9: Quality risk management; 2005. https://database.ich.org/sites/default/files/Q9_Guideline.pdf Accessed 01 July 2022.
World Health Organisation (WHO), Annex 11, Good regulatory practices in the regulation of medical products. Fifty fifth report. https://www.wto.org/english/tratop_e/trips_e/techsymp_290621/gaspar_pres2.pdf. Accessed 04 July 2022.
World Health Organisation (WHO). A framework for risk-based identification of essential medicine products for local manufacturing in low- and middle-income countries, Concept paper, WHO Drug Information Vol. 30, No. 1, 2016.
Cone M, McAuslane N. The Centre for innovation in Regulatory Science, Building Quality into Regulatory Dossiers and the Review Process, R&D Briefing 46, Novellus Court, 61 South Street, Epsom, UK, 2006.
https://www.cirsci.org/publications/cirs-rd-briefing-46-building-quality-into-regulatory-activities/. Accessed 26 July 2022.
Medicine Control Council (MCC) archives. MCC 77 presentation; Backlog status, dated: 21-22 April 2016.
World Health Organisation (WHO) Drug Information, Towards a global competency framework for regulators of medical products. Regulatory News. 2019;33:6-9.
Yu XL, Raw A, Lionberger R. US FDA question-based review for generic drugs: A new pharmaceutical quality assessment system. J Gen Med. 2007;4(4):239–48.
Bujar M, Patel P, McAuslane N, Liberti L. Centre for Innovation in Regulatory Science (CIRS). The changing regulatory environment in Latin America, Focus on good review practices, R&D Briefing 58, The Johnson Building, 77 Hatton Garden London, UK, 2015.
Lilly website. Benefit-risk balance of medicines, https://www.lilly.com/medicines/safety/benefit-risk-balance Accessed 22 June 2022.
Keyter A, Salek S, Banoo S, Walker S. A proposed regulatory review model to support the south African health products regulatory authority to become a more efficient and effective agency. Int J Health Policy Manag. 2020. https://doi.org/10.34172/ijhpm.2020.213.
Moeti L, Litedu M, Joubert J. Common deficiencies found in generic finished pharmaceutical products (FPPs) submitted for registration by the South African Health Products Regulatory Authority (SAHPRA). J Pharm Pol Prac. 2022;15:1–21.
Leong J, Salek S, Walker S. Benefit-risk assessment of medicines; The Development and Application of a Universal Framework for Decision-Making and Effective Communication, Springer International Publishing, Switzerland, 2015.
World health organisation prequalification Team – medicines website: List of prequalified APIs and FPPs. https://extranet.who.int/prequal/content/prequalified-lists/medicines Accessed 25 June 2022.
European Directorate for the Quality of Medicines. Certification of suitability to Monographs of the European Pharmacopoeia: https://www.edqm.eu/en/certificate-suitability-new-applications Accessed 01 March 2022.
Gosar A, Sayyed H, Shaikh T. Genotoxic impurities and its risk assessment in drug compounds. Drug Des Int Prop Int J. 2018. https://doi.org/10.32474/DDIPIJ.2018.02.000143.
Tran NL, Hasselbalch B, Morgan K, Claycamp G. Elicitation of Expert knowledge about risk associated with pharmaceutical manufacturing processes. Pharmac Eng. 2005;25(4):1–12.
Censi R, Martino PD. Polymorph Impact on the bioavailability and stability of poorly soluble drugs. Mol. 2015. https://doi.org/10.3390/molecules201018759.
Sam T, Ernest T, Walsh J, Williams J. A benefit/risk approach towards selecting appropriate pharmaceutical dosage forms—an application for paediatric dosage form selection. Int J Pharm. 2012. https://doi.org/10.1016/j.ijpharm.2012.05.024.
Zammit M. Risk Management in the Manufacture of Solid Oral Dosage Forms, University of Malta, Department of Pharmacy, 2017. Available: https://www.researchgate.net/publication/334634509_Risk_Management_in_Manufacture_of_Solid_Oral_Dosage_Forms. Accessed 17 June 2022.
Gordon J, Stahl M, Lembit R, Potthast H. World Health Organisation (WHO). 2017; DOI: https://doi.org/10.1007/978-3-319-68078-1_11, ISBN 978-3-319-68077-4.
Manufacture of sterile medicinal products. In: The rules governing medicinal products in the European Union Vol. 4. EU guidelines to good manufacturing practice medicinal products for human and veterinary use. Annex 1, Brussels, 2008. Available: https://health.ec.europa.eu/latest-updates/eudralex-volume-4-eu-guidelines-good-manufacturing-practice-medicinal-products-human-and-veterinary-2022-02-21_en. Accessed 25 June 2022.
Akers MJ. Sterile Drug Products: Formulation, Packaging, Manufacturing and Quality, Informa Healthcare, New York, USA, 2010.
Riley B, Li X. Quality by design and process analytical technology for sterile products where are we now. AAPS Pharm Sci Tech. 2011;12:114–8.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Biopharmaceutics Classification System-Based Biowaivers M9, 20 November 2019. https://database.ich.org/sites/default/files/M9_Guideline_Step4_2019_1116.pdf. Accessed 25 June 2022.
United States Food and Drug Administration, Quality by Design for ANDAs: An Example for Immediate-Release Dosage Forms, Pharmaceutical Development Report, April 2012. https://www.fda.gov/files/drugs/published/quality-by-design-%28QbD%29-for-an-immediate-release.pdf. Accessed 02 July 2022.
Gupta KR. Role of Environmental Monitoring and Microbiological Testing during Manufacture of Sterile Drugs and Biologics, American Pharmaceutical Review, European Microbiology Conference held on 7-8 May 2014 at Prague, Czech Republic. May 2014. Available: https://www.americanpharmaceuticalreview.com/Featured-Articles/169384-Role-of-Environmental-Monitoring-and-Microbiological-Testing-During-Manufacture-of-Sterile-Drugs-and-Biologics/. Accessed 14 June 2022.
European Guidelines to Good Manufacturing Practice. Medicinal Products for Human and Veterinary Use, Volume 4, Annex 1, Manufacture of Sterile Medicinal Products, 2008. http://ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf. Accessed 27 May 2022.
Pharmaceutical Inspection Convention (PIC/S). Guide to Good Manufacturing Practice for Medicinal Products. Annex 1, Manufacture of Sterile Medicinal Products, 2014. Available: https://picscheme.org/docview/4590 Accessed 27 May 2022.
European Medicines Agency, Committee for Medicinal Products for Human use (CHMP), Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container EMA/CHMP/CVMP/QWP/850374/2015, 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-sterilisation-medicinal-product-active-substance-excipient-primary-container_en.pdf Accessed 05 July 2022.
European Medicines Agency. Committee for Medicinal Products for Human use (CHMP), Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container EMA/CHMP/CVMP/QWP/850374/2015, 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-sterilisation-medicinal-product-active-substance-excipient-primary-container_en.pdf. Accessed 05 July 2022.
Drug Bank Online. Narrow therapeutic index drugs. Available: https://go.drugbank.com/categories/DBCAT003972 Accessed 05 July 2022.
Tomlinson C. The plan to revive medicines regulation in South Africa, Spotlight, 2019. Available: The plan to revive medicines regulation in South Africa • Spotlight (spotlightnsp.co.za). Accessed 13 October 2022.
South African Health Products Regulatory Authority (SAHPRA). Backlog clearance programme, extension of project, Industry communication, 22 June 2022. https://www.sahpra.org.za/wp-content/uploads/2021/06/Backlog-Clearance-Programme_Extension-of-Project_22.06.2021_vF.docx.pdf. Accessed 06 October 2021.
South African Health Products Regulatory Authority (SAHPRA). Reliance guideline, March 2022. Available: https://www.sahpra.org.za/wp-content/uploads/2022/03/Reliance-Guideline_v3_22-Mar-2022.pdf Accessed 07 October 2022.
International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). Structure and content of clinical study reports, 30 November 1995. https://database.ich.org/sites/default/files/E3_Guideline.pdf. Accessed 21 June 2022.
European Medicines Agency. Committee for Medicinal Products for Human use (CHMP), Guideline on the investigation of bioequivalence, CPMP/EWP/QWP/1401/98 Rev. 1, 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 03 March 2022.
Acknowledgements
The following are greatly acknowledged for their tireless efforts and contribution in the backlog Phase 1 pilot project, 2015–2016 and the 2021 pilot study, Phase 1: Dr Mbali Keele who was part of the team involved in the development and leadership of the two backlog phases in 2015–2016. Dr Joey Gouws for the immense support in the drive of the backlog pilot study in 2016. The backlog working group, which consisted of all the representatives from all the units responsible for registration of human medicines within MCC for the efforts and inputs in the success of the pilot backlog project. Ms Lorraine Danks as the head of the Backlog Clearance Programme, for the immense support and championship of the risk-based assessment pilot study in 2021. Dr Boitumelo Semete-Makokotlela, Ms Portia Nkambule and Ms Christelna Reynecke for their full support in the drive and implementation of the backlog pilot study Phase 1, 2021. All the evaluators who took part in the two risk-based assessment pilot studies for their contributions to the success of the projects. Ms Joy Van Oudtshoorn for assistance in the development of some of the templates used in the pilot study in 2021. Prof. Theo Dekker and Prof. Admire Dube for their valuable input on the risk classification matrices used and support in the refinement of the project for implementation by SAHPRA. The technical advisory working group (TAWG) for the valuable input in the proposed process of the pilot study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The open-access publication fee for this research was supported by an unrestricted grant from the Bill and Melinda Gates Foundation.
Conflicts of interest
The authors have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data are available upon request to the corresponding author.
Code availability
Not applicable.
Author contributions
LM: Developed the study design, collected and analysed the data, interpreted the results, and wrote the first draft of the manuscript. ML: Developed the study design, assisted in collecting and analysing the data, provided guidance for the data collection and analysis, interpreted the results, and reviewed and approved the manuscript. JJ: Developed the study design, provided guidance on the data analysis, interpretation and relevance of the results, and reviewed and approved the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Moeti, L., Litedu, M. & Joubert, J. The Implementation of a Risk-Based Assessment Approach by the South African Health Products Regulatory Authority (SAHPRA). Pharm Med 37, 71–91 (2023). https://doi.org/10.1007/s40290-022-00452-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-022-00452-w