
Abstract
The physiological determinants of high-intensity exercise tolerance are important for both elite human performance and morbidity, mortality and disease in clinical settings. The asymptote of the hyperbolic relation between external power and time to task failure, critical power, represents the threshold intensity above which systemic and intramuscular metabolic homeostasis can no longer be maintained. After ~ 60 years of research into the phenomenon of critical power, a clear understanding of its physiological determinants has emerged. The purpose of the present review is to critically examine this contemporary evidence in order to explain the physiological underpinnings of critical power. Evidence demonstrating that alterations in convective and diffusive oxygen delivery can impact upon critical power is first addressed. Subsequently, evidence is considered that shows that rates of muscle oxygen utilisation, inferred via the kinetics of pulmonary oxygen consumption, can influence critical power. The data reveal a clear picture that alterations in the rates of flux along every step of the oxygen transport and utilisation pathways influence critical power. It is also clear that critical power is influenced by motor unit recruitment patterns. On this basis, it is proposed that convective and diffusive oxygen delivery act in concert with muscle oxygen utilisation rates to determine the intracellular metabolic milieu and state of fatigue within the myocytes. This interacts with exercising muscle mass and motor unit recruitment patterns to ultimately determine critical power.
Similar content being viewed by others
Critical power represents the threshold intensity above which steady-state metabolism is no longer attainable, and within the last ~ 15 years, experimental data have emerged that illuminate its underpinning physiological determinants. |
Here, we summarise these experimental data to demonstrate that critical power is a parameter of aerobic function that is affected by alterations in the capacities of each step in the oxygen transport and utilisation pathways. |
Convective/diffusive oxygen delivery and intracellular oxygen utilisation rates interact with muscle fibre composition and motor unit recruitment profiles to determine the upper limit for steady-state exercise. |
1 Introduction
The determinants of exercise tolerance are of clear interest because of the strong relationships between exercise capacity and athletic performance [1, 2], health in the general population, and clinical outcomes in disease populations [3, 4]. Exercise intensity is, of course, a key factor that determines the tolerability of a given task. Moreover, for individuals or groups of individuals, partitioning the exercise intensity spectrum into domains where the physiological responses to a given task share common qualitative characteristics is an effective approach that can yield insight into the physiological determinants of exercise tolerance. Accordingly, the mechanisms of fatigue and determinants of exercise intolerance are not ubiquitous across the spectrum of exercise intensities [5]. However, above a particular individual-specific power output, the consistent feature of exercise intolerance (and hence, impending task failure) is the inability for pulmonary oxygen uptake (\(\dot{V}\)O2) and [lactate] (L−) to attain a steady state [6,7,8,9]. Thus, for each individual, there exists a range of intensities for which a steady state in pulmonary \(\dot{V}\)O2 is attainable, and a range for which it is not [6, 9,10,11,12], with the duration of sustainable exercise in the latter being significantly limited compared with the former. The threshold intensity that separates these two ranges of system behaviour, and its position relative to other landmarks of aerobic function (i.e. maximal \(\dot{V}\)O2 [\(\dot{V}\)O2max] and the lactate threshold), is therefore a fundamental determinant of the ability to sustain exercise [6, 13,14,15].
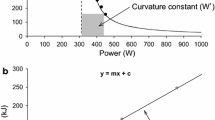
This threshold intensity can be determined by undertaking three to five high-intensity, constant-power output cycle ergometer tests to the point of task failure on separate days. The tests should be selected to last no less than 2 and no more than 15 min in duration [16,17,18,19], with the precise time to task failure and power output at which each test is conducted recorded. These durations are recommended for a valid determination of this intensity, as it is essential that \(\dot{V}\)O2max is attained at the end of trial in order to meet the requirement for all prediction trials to be performed within the severe-intensity domain. When time to task failure is plotted against power output, the relationship is curvilinear, with the ability to sustain exercise falling away more rapidly at higher power outputs (Fig. 1). This power-time relationship is well described by a hyperbolic function [20], with an asymptote known as critical power (CP) and the curvature constant termed W' (i.e. W prime). This relationship is described by the following equation:
where T is the tolerable duration and P is the power output of a given exercise task [6, 20, 21]. When intensity is measured in units of speed, the asymptote is termed critical speed (CS) and the curvature constant D’ (i.e. with units of distance). This power-time relationship appears to be a universal feature of high-intensity exercise tolerance, being apparent in every species [22,23,24,25,26] and mode of exercise (with appropriate units of force, torque or velocity [15, 27,28,29,30]) in which it has been studied. This relationship can also be converted to its linear equivalents, either with work plotted against time:
where W is work, CP is the slope and W′ is the intercept of the equation, or with power plotted against the inverse of time:
or
where CP is the intercept and W' is the slope of the equation.

Hyperbolic power-duration curve that defines the sustainable duration of exercise in the severe-intensity domain. This hyperbolic relationship is defined by two parameters: the power asymptote, known as the critical power (CP), and the curvature constant W′ (denoted by the rectangular dashed blue lines above CP and expressed in kilojoules). Critical power defines the boundary between the heavy- and severe-intensity exercise domains and represents the highest power output for which a metabolic steady state may be attained. The W' comprises a fixed and finite volume of work that is expendable above CP. During severe-intensity exercise, task failure occurs when W′ = 0. GET gas exchange threshold
Since the seminal work by Prof. David Poole and colleagues in the late 1980s, it has been repeatedly demonstrated that CP reflects the upper limit at which a metabolic steady state can be sustained. The basis for this has been the ubiquity of steady-state behaviour of metabolic variables associated with aerobic function below, but not above, CP. For example, \(\dot{V}\)O2 rises to \(\dot{V}\)O2max during exercise above, but not at or below, CP [6], accompanied by similarly inexorable trajectories of blood [lactate], [HCO3−] and pH [6, 31]. Such findings were subsequently confirmed in different populations, including the elderly [32], patients with chronic heart failure [33] and patients with chronic obstructive pulmonary disease (COPD) [34, 35], and healthy children [36]. More recently, non-invasive (31P-magnetic resonance spectroscopy, near-infrared spectroscopy) and invasive (i.e. muscle biopsy) studies have demonstrated the achievement of a steady state in the exercising muscle below, but not above, CP in muscle \(\dot{V}\)O2, [phosphocreatine] ([PCr]), [inorganic phosphate] [Pi], pH and muscle [lactate] [15, 31, 37]; for a review, see [8, 11, 12]. Critical speed (an analogue of CP) has also been demonstrated to be a critical threshold for motor unit recruitment patterns, with Copp et al. demonstrating that exercise above CS was accompanied by disproportionate increases in blood flow to type IIb/d/x fibres in the rat hind limb muscle [25].
Despite CP, and its analogues of external expression (i.e. critical speed, torque, force) being widely recognised as reflecting the threshold intensity above which a metabolic steady state cannot be sustained, its physiological antecedents have previously been obscure. Tables 1 and 2 detail interventional and observational approaches to understanding CP. Prior to the year 2010, intervention studies on CP were scant, and primarily confined to the effect of exercise training alongside additional measures of \(\dot{V}\)O2max and the gas exchange threshold/lactate threshold only, although one of the earliest studies on CP did show an independent effect of O2 availability on CP (albeit in just two participants [21]). Nevertheless, such findings supported the notion of CP as a parameter of aerobic function [20]. In contrast, since 2010, multiple experimental approaches have revealed those factors that, directly or indirectly, determine CP. The purpose of the present review is therefore to examine the physiological and biochemical underpinnings of this fundamental parameter of exercise tolerance. Particular attention will be paid to evidence generated over the last 10–12 years demonstrating that CP is a key parameter of aerobic function that can be affected by any step in the O2 transport and utilisation pathway.
2 Interaction of Factors Determining CP
That CP represents the threshold intensity above which exercise cannot be sustained in a steady state indicates that it is a parameter of aerobic function. Consequently, it follows that CP may be affected by any step in the O2 transport and utilisation cascade, from atmospheric air down to the muscle mitochondria themselves. Specifically, these steps include: (1) transport of atmospheric O2 into the blood via pulmonary diffusion; (2) bulk transport of O2 to the muscle via convection (i.e. convective O2 delivery); (3) diffusion of O2 from capillary to muscle mitochondria (i.e. diffusive O2 delivery); and (4) the utilisation of O2 by the muscle mitochondria (Fig. 2). Whilst the respiratory system may constrain CP in chronic respiratory disease conditions such as COPD [34, 35, 38,39,40,41,42], in most young healthy individuals, the respiratory system appears to be well adapted to ensure a highly efficient and appropriate homeostatic response to high-intensity exercise [43]. Hence, the remainder of this review will focus on the impact of convective and diffusive O2 delivery and mitochondrial O2 utilisation on CP, downstream of the respiratory system.

Adapted from Wasserman et al. [156], with permission
Schematic illustrating an adaptation of Wasserman’s classic “Gears” diagram. It demonstrates Wasserman’s conception of how the respiratory, cardiovascular and neuromuscular systems conflate to enable exercise to be sustained. O2 flows from the atmosphere through the lungs, pulmonary and peripheral circulation to the muscle mitochondria where it is ultimately consumed. CO2 produced by the contracting muscle flows along the same pathway in reverse. Muscle work leads to increased cardiac output and redistribution of blood flow, and increased ventilation in response to both the increased metabolism and evolution of CO2 from the blood as the result of lactic acid buffering. The efficacy of these processes determines the ability to sustain muscular exercise. These concepts are reconsidered in this review within the context of critical power. This figure was created with BioRender.com and was exported under a paid subscription. \(\dot{Q}\)CO2 cellular carbon dioxide production, \(\dot{Q}\)O2\(/\dot{V}\)O2 matching matching of oxygen delivery to local oxygen consumption, \(\dot{Q}\) O2 cellular oxygen consumption, \(\dot{Q}/\dot{V}\) matching matching of ventilation to perfusion, \(\dot{V}\)A alveolar ventilation, \(\dot{V}\mathrm{CO}2\) Pulmonary carbon dioxide production, \(\dot{V}\)D dead space ventilation, \(\dot{V}\)E minute ventilation, \(\dot{V}\)O2 pulmonary oxygen consumption.
2.1 Convective Oxygen Delivery
Convective O2 delivery refers to that achieved via bulk movement of O2 within the circulation to the exercising muscles. Convective O2 delivery (\(\dot{Q}\)O2, L min−1) can thus be defined mathematically as the product of cardiac output (CO, L min−1) and arterial O2 content (CaO2, mL O2 100 mL−1):
where CaO2 is defined as:
where 1.34 is Hüfner’s constant describing the maximum O2 carrying capacity per gram of haemoglobin (mL O2 g−1 Hb), [Hb] is haemoglobin concentration (g dL−1), SaO2 is the arterial saturation of Hb, 0.03 is the solubility coefficient of O2 at body temperature (mL O2 100 mL−1 plasma kPa−1) and PaO2 is the arterial partial pressure of O2 (mmHg). This provides a measure of whole-body convective O2 delivery. However, the O2 flux to each portion of the exercising muscles is not uniform but varies according to regional metabolic demands, vascular control and fibre type [44,45,46].
A convenient means by which to experimentally alter CaO2, and hence, convective O2 delivery, is by varying the fraction of inspired O2 (FiO2). Although hypoxia-induced vasodilatation [47] and hyperoxia-induced vasoconstriction [48] often influence blood flow thereby helping to normalise muscle O2 delivery during exercise, many studies that have quantified skeletal muscle O2 delivery under these conditions have demonstrated that hyperoxia can enhance and hypoxia can impair skeletal muscle O2 delivery during exercise, respectively [49,50,51,52,53,54], thereby impacting upon intra-myocyte PO2 (PO2im) [55]. Indeed, the early work of Moritani et al. [21] showed that, in a limited sample of two participants, inspiration of a hypoxic gas mixture (FiO2 0.09) resulted in a reduced CP compared with normoxia (i.e. FiO2 0.21; hypoxia: 106 ± 6 W, vs normoxia: 214 ± 4 W). Under conditions of more moderate hypoxia (FiO2 0.15) and in a larger sample of 11 subjects, Dekerle et al. [56] found that CP was reduced by 30 W in hypoxia compared with normoxia, consequent to a reduction in arterial O2 saturation of 12%. Notably, in this latter study, the percentage decrement in CP between hypoxia and normoxia was correlated with \(\dot{V}\)O2max in normoxia, suggesting that those with the greatest \(\dot{V}\)O2max values were better able to offset the reductions in convective O2 delivery brought about via hypoxia. It is not known if such a protective effect remains in highly trained athletes where pulmonary limitations to high-intensity exercise are more likely [57, 58], causing reductions in arterial saturation and \(\dot{V}\) O2max even at modest simulated altitudes [59]. Similarly, however, Simpson et al. [60] reported a reduction in CP of 43 W using an FiO2 of 0.13, a finding that was consistent when CP was determined either via the conventional constant-load prediction trial method or via a 3-min all-out test. Moreover, Valli et al. [61] demonstrated that at an altitude of 5050 m (equivalent FiO2 ~ 0.11) CP was reduced by 42 W. In all of these studies, SaO2 was reduced either at rest or during exercise in hypoxia, providing indirect evidence that hypoxia impaired convective O2 delivery that contributed to the reduced CP in each study. These findings were subsequently extended to arm cycle ergometry by La Monica et al. [62], who demonstrated that arm CP was reduced by 5 W in moderate (FiO2 0.14) normobaric hypoxia (~ 6% of normoxic CP). Whilst the magnitude of the effect of hypoxia on CP in these studies varied with the fitness of the participants (see, for example, Dekerle et al. [56]), Townsend et al. [63] demonstrated a progressive reduction in CP with decreasing FiO2. Hence, the extant literature is unanimously consistent with the notion that reductions in FiO2 (and by extension, convective O2 delivery) reduce CP.
The consistency of the effects of hyperoxia on CP are similar to those of hypoxia. This was first demonstrated by Vanhatalo et al. [37], who assessed the impact of an FiO2 of 0.7 on CP utilising a single-leg knee-extension exercise model. These authors showed that CP was increased in hyperoxia compared with normoxia, with a concomitant increase in muscle oxygenation (as determined via near-infrared spectroscopy [NIRS]). The increase in CP was accompanied by a slower rate of change in muscle [PCr], [ADP], [Pi] and pH. Subsequently, these findings for small-muscle mass exercise were confirmed for large-muscle mass exercise by Goulding et al. [64, 65]. Specifically, a hyperoxic inspirate (FiO2 of 0.5) resulted in increases in end-tidal PO2 (and, therefore, alveolar PO2) and muscle oxygenation determined via NIRS both at rest and during exercise [64, 65]. As a result, CP was enhanced during cycle exercise in hyperoxia versus normoxia in both the supine [64] and upright [65] body positions, with the magnitude of improvement being ~ 10% in both studies. Hence, studies have consistently shown that CP is sensitive to both increased [37, 64, 65] and decreased [21, 56, 60,61,62] FiO2.
Another experimental intervention that has yielded insights into the dependency of CP on convective O2 delivery is via manipulations in the muscle contraction duty cycle. Muscle contraction, particularly during small-muscle mass exercise where compressive forces can be high, increases intramuscular pressure, compresses blood vessels, increases impedance to flow and may cause temporary blood flow occlusion [66,67,68,69]. Hence, the muscular contraction cycle yields rhythmic alterations in intramuscular pressure, and hence blood flow, with the majority of flow occurring during the relaxation phase of contraction, [69,70,71,72]. Utilising a small-muscle mass handgrip exercise, Broxterman et al. [73] directly tested the hypothesis that alterations in the duty cycle would cause concomitant alterations in convective O2 delivery, and hence CP, by measuring brachial artery blood flow via Doppler ultrasound during exercise with a 20% and 50% duty cycle (i.e. muscle contraction comprised 20 and 50%, respectively, of the total contraction-relaxation cycle). Brachial artery blood flow, and thus, convective O2 delivery, was greater in the 20% duty cycle when compared with the 50% duty cycle, with a concomitant increase in CP [73].
In extending the principle of altering convective O2 delivery to observe its effect on CP, Broxterman et al. [74, 75] showed that during blood flow occlusion (which constrains O2 delivery to zero), CP was reduced to a negative value. Whilst a negative CP appears implausible, this finding demonstrates a reliance of CP on convective O2 delivery as there is no sustainable rate of oxidative metabolism without blood flow. Resting (i.e. 0 W) occlusion results in progressive depletion of [PCr] and muscle/capillary O2 stores [76, 77], a feature consistent with non-steady-state conditions [15]. Accordingly, the magnitude of the negative CP during blood flow occlusion would be expected to be proportional to the resting metabolic rate, and as such is entirely plausible.
These findings were recently extended by Hammer et al. [78] where critical force (CF) was estimated during the final minute of repeated handgrip maximum voluntary contraction (MVC) efforts over a 5-min duration. Under free-flowing conditions without occlusion, force progressively declined with time during the test until a plateau was reached in the final minute of the test, termed CF [78]. With muscle occlusion, however, force continuously declined with time, i.e. there was no plateau in force at the end exercise [78]. Following subsequent reperfusion, force was able to recover to a level not significantly different from CF determined under free-flowing conditions [78]. These authors also demonstrated that up to and including CF, end-exercise limb blood flow values were linearly related to the constant-force requirements of each task [79, 80]. However, during exercise slightly above CF, end-exercise brachial artery blood flow demonstrated a plateau, being no different from the blood flow values obtained during exercise at CF [79]. These findings were subsequently extended to large muscle mass, whole-body exercise by the same authors [80]. Specifically, leg blood flow and limb vascular conductance were determined using Doppler ultrasound and calibrated finger plethysmography during exercise above and below CP [80]. Post-exercise increases in limb vascular conductance and leg blood flow post-exercise were observed following supra-CP but not sub-CP exercise [80]. The data of Hammer et al. [79, 80] are in contrast to observations in the running rat [25] and from upright, incremental, large muscle mass exercise in humans [52, 81] showing increases in limb blood flow up to \(\dot{V}\)O2max. Nevertheless, these findings raise the intriguing possibility that in certain contexts, CF/CP represents a threshold in relative muscular force that limits skeletal muscle perfusion during exercise. Moreover, the extant literature appears to be unanimously consistent with CP being determined, at least in part, by mechanisms related to convective O2 delivery.
2.2 Diffusive Oxygen Transport
Diffusive O2 transport refers to the diffusive movement of O2 from the capillaries to the muscle mitochondria where O2 serves as the final electron acceptor for the electron transport system. This process is described mathematically via Fick’s law of diffusion:
where \(\dot{V}\)O2 corresponds to the rate of O2 flux, DO2 is the muscle diffusing capacity, and ΔPO2 is the partial pressure difference between the capillary and intra-myocyte spaces (PO2cap and PO2im, respectively). This relationship dictates that elevations in \(\dot{V}\)O2 must be established via changes in either (1) changes in the driving force for O2 diffusion (i.e. ΔPO2 = PO2cap − PO2im) and/or (2) changes in effective diffusing capacity (i.e. DO2, determined primarily by the aggregate number of blood cells within capillaries adjacent to the myocyte at any given moment [82, 83]).
Fick’s Law of Diffusion predicts that alterations in FiO2 will bring about concomitant alterations in CP via altered O2 diffusion in addition to convection. For instance, hypoxia reduces and hyperoxia increases both estimated PO2cap [84] and PO2im [85], though to differing extents such that ∆PO2 is reduced and increased, respectively. Hence, in the studies reviewed in Sect. 2.1 wherein hypoxia reduced [21, 56, 60,61,62] and hyperoxia increased CP [37, 64, 65], it is also probable that alterations in the transcapillary driving force for O2 flux, and thus diffusive O2 delivery, also contributed to the alterations in CP observed therein, likely via the alterations this would be expected to have on PO2im [55].
Muscle capillarity is an important influence on DO2, and thus diffusive O2 delivery, as it determines the number of red blood cells adjacent to contracting fibres and thus the surface area available for O2 diffusion. Indeed, Mitchell et al. [86] recently demonstrated a striking relationship between CP and skeletal muscle capillary density (r = 0.50), capillary-to-fibre ratio (r = 0.88) and capillary contacts per type 1 fibre (r = 0.94) in a homogenous group of endurance-trained individuals (63.2 ± 4.1 mL kg−1 min−1, range: 58.7–72.2 mL kg−1 min−1). These findings indicate that enhancements in diffusive O2 flux enable a metabolic steady state to be attained for a greater range of power outputs (i.e. extending the range upwards), thus increasing CP.
Further insight into the role of diffusive factors in determining CP/CF was provided by a series of experiments by Ansdell et al. that compared the power-duration relationship between the sexes during small- [87] and large-muscle mass exercise [88]. It was demonstrated that CF occurred at a greater relative percentage of the MVC in female individuals compared with male individuals during small-muscle mass, intermittent, isometric single-leg knee extension exercise [87]. Conversely, there were no differences observed in the relative percentage of MVC at which CP occurred between male and female individuals during large-muscle mass dynamic cycle exercise [88]. Female individuals have previously been demonstrated to possess a greater degree of capillarity in skeletal muscle and a greater proportion of type I fibres when compared with male individuals [89,90,91], suggesting a greater capacity for diffusive O2 transport. Moreover, during small-muscle mass knee extension exercise, far greater mass-specific rates of blood flow are achieved when compared with cycle exercise, and hence, diffusive rather than convective factors constrain O2 transport to muscle mitochondria [52, 81, 92,93,94,95,96,97]. These authors [87, 88] consequently interpreted their findings to indicate that during single-limb exercise where convective factors are not limiting, the sex difference in CF arises because of a greater skeletal muscle diffusive capacity of female individuals [87, 88]. Conversely, during dynamic cycle exercise where muscle O2 delivery is constrained by the central nervous system to prevent a dangerous fall in mean arterial pressure [98], convective O2 delivery may be relatively more important in determining CP than muscle diffusive capacity, leading to the lack of a sex difference in this mode of exercise [87, 88].
Utilising measurements of brachial artery blood flow via Doppler ultrasound and NIRS to determine muscle O2 extraction, Broxterman et al. [73] were able to estimate muscle \(\dot{V}\)O2 and thereby estimate the contributions of enhanced convective and diffusive O2 delivery to the changes in CP they observed between 20 and 50% duty cycles (discussed in Convective Oxygen Delivery). These authors demonstrated that the increase in DO2 in the 20% versus the 50% duty cycle was approximately double the increase in convective O2 delivery that occurred between the same trials (i.e. + 69% vs + 34%, respectively), implicating changes in diffusive, rather than convective, O2 delivery as being a more important determinant of CP in this situation. These authors suggested that the shorter duty cycle would have facilitated higher red blood cell velocity and therefore increased the surface area of the capillary involved in gas exchange (i.e. longitudinal capillary recruitment [99]), thereby enhancing DO2 and contributing to the increased CP. Interestingly, this observation is also consistent with the suppositions of Ansdell et al. [87, 88] noted above, namely that diffusive factors may be more important for constraining CP during small- versus large-muscle mass exercise. That DO2 is an independent determinant of CP was recently confirmed by Colburn et al. [100]. Specifically, the vascular ATP-sensitive K+ channel inhibitor glibenclamide decreased CS in rats, and this was accompanied by a 25% decrease in DO2 determined from measurements of skeletal muscle blood flow, arterial O2 content, and interstitial and microvascular O2 pressures [100]. Collectively, therefore, there is now a growing body of evidence to indicate that CP can be influenced by factors dictating the rate of diffusion of O2 from capillaries to mitochondria.
2.3 Oxygen Utilisation
A sentinel parameter defining the skeletal muscle bioenergetics system is the time constant of the fundamental phase of muscle \(\dot{V}\)O2 kinetics (i.e. \({\tau }_{\dot{V}\mathrm{O}2}\)), which is reflective of the time taken to attain 63% of the \(\dot{V}\)O2 amplitude in response to a change in metabolic demand [101,102,103,104], and is closely reflected by the pulmonary \({\tau }_{\dot{V}\mathrm{O}2}\) [103]. Pulmonary \({\tau }_{\dot{V}\mathrm{O}2}\) is therefore a highly convenient assay of the time course of changes in oxidative phosphorylation that occur at the onset of exercise or during changes in the metabolic rate. At the onset of exercise, therefore, the delayed response of pulmonary and muscle \(\dot{V}\)O2 kinetics that is encapsulated by the parameter \({\tau }_{\dot{V}\mathrm{O}2}\) necessitates an energy deficit that must be met via a reduction in O2 stores and an increased rate of substrate-level phosphorylation [103, 105, 106]. This “O2 deficit” is a function of \({\tau }_{\dot{V}\mathrm{O}2}\) and the steady-state increment \(\dot{V}\)O2 [105], at least for work rates where a steady state is rapidly attained. The magnitude of this O2 deficit at exercise onset is critical, as it determines (1) the degree of reliance on non-oxidative sources of energy provision (i.e. depletion of [PCr] and [glycogen] and consequent accumulation of [L−] and [H+]), (2) the magnitude of metabolic perturbation incurred during the rest-to-work transition (i.e. Δ[PCr], Δ[ADP], Δ[Pi], extracellular [K+] accumulation, loss of sarcoplasmic Ca2+ release and sensitivity), (3) the extent of fatigue induction sustained and (4) the loss of skeletal muscle efficiency induced during the rest-to-exercise transition [8, 10, 14, 101, 102, 104, 107,108,109,110]. \(\dot{V}\)O2 kinetics would therefore appear to be central in setting the tolerability of exercise. Indeed, very low \({\tau }_{\dot{V}\mathrm{O}2}\) values (i.e. fast \(\dot{V}\)O2 kinetics) are observed in endurance athletes [111] and trained individuals [112], whereas very large \({\tau }_{\dot{V}\mathrm{O}2}\) values (i.e. slow \(\dot{V}\)O2 kinetics) are observed in the elderly [113] and chronically ill [102]. However, until relatively recently, an independent role for \({\tau }_{\dot{V}\mathrm{O}2}\) in determining CP had not been considered.
Murgatroyd et al. [14] characterised relationships between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP by normalising exercise intensity across individuals such that the tolerable duration of exercise was uniform (6 min). They demonstrated a strong inverse correlation between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP (r = 0.95), consistent with the notion that \({\tau }_{\dot{V}\mathrm{O}2}\) has an independent role in determining CP. Moreover, when this analysis was extended across human populations spanning the extremes of aerobic function (i.e. healthy young trained individuals, young inactive individuals, healthy elderly individuals and patients with COPD), the relationship between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP was strong, inverse and linear [104]. These authors interpreted this relationship causally: by minimising the reliance on substrate-level phosphorylation, and hence the accumulation of fatigue-related metabolites during the transition, a lower \({\tau }_{\dot{V}\mathrm{O}2}\) (i.e. faster \(\dot{V}\)O2 kinetics) allows a higher power production to be achieved for a given magnitude of O2 deficit accumulation. Critical power represents the upper limit of the metabolic steady state, and by extension also signifies the upper limit of an O2 deficit below which muscle fatigue, reduction in work efficiency and the O2 deficit itself will stabilise. All else being equal, therefore, faster \(\dot{V}\)O2 kinetics will result in a higher CP. However, despite the strong rationale and cross-sectional evidence supporting a mechanistic link between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP, until recently, this hypothesis had not received direct experimental scrutiny.
In the first of a series of studies examining the purported determining effect of \({\tau }_{\dot{V}\mathrm{O}2}\) on CP, Goulding et al. [114] examined the influence of prior heavy (“priming”) exercise on pulmonary \(\dot{V}\)O2 kinetics and CP during supine and upright cycling. A prior bout of priming exercise does not speed \(\dot{V}\)O2 kinetics (i.e. reduce \({\tau }_{\dot{V}\mathrm{O}2}\)) during upright cycle exercise in young healthy individuals. However, during exercise in the supine position, muscle perfusion pressure is impaired and \({\tau }_{\dot{V}\mathrm{O}2}\) becomes O2 delivery dependent [114,115,116,117,118,119,120]. Hence, in a young healthy population, prior heavy exercise (which enhances muscle O2 delivery, [115, 121, 122]) would be expected to reduce \({\tau }_{\dot{V}\mathrm{O}2}\) during supine but not upright cycling. Accordingly, should \({\tau }_{\dot{V}\mathrm{O}2}\) exert a determining effect on CP, an increase in CP during supine, but not upright, exercise would be observed following priming exercise as compared with control conditions. It was demonstrated that when priming exercise was conducted in the supine position, \({\tau }_{\dot{V}\mathrm{O}2}\) was indeed reduced and CP concomitantly increased, whereas during upright exercise, both \({\tau }_{\dot{V}\mathrm{O}2}\) and CP were unaffected [114]. These findings therefore provided the first experimental evidence that \({\tau }_{\dot{V}\mathrm{O}2}\) is mechanistically related to CP.
Because of the nature of the priming intervention utilised in this first study [114], however, it was not possible to separate any independent effect of a reduced \({\tau }_{\dot{V}\mathrm{O}2}\) (i.e. slowed \(\dot{V}\)O2 kinetics) on CP from that of an improved O2 availability as a consequence of the priming exercise. Indeed, the strong correlation observed between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP for upright exercise was absent for supine exercise [114]. Hence, it remained plausible that, at least in supine exercise, other physiological factors, such as muscle O2 availability, and its distribution relative to \(\dot{V}\)O2, determine CP, with the concomitant improvements in \({\tau }_{\dot{V}\mathrm{O}2}\) and CP being an artefact of shared physiological determinants, without any dependence of CP on \({\tau }_{\dot{V}\mathrm{O}2}\) per se. Hence, confirmation or refutation of the hypothesis that \({\tau }_{\dot{V}\mathrm{O}2}\) is an independent determinant of CP required an intervention that could alter \({\tau }_{\dot{V}\mathrm{O}2}\) without any concomitant alterations in muscle O2 delivery, such that the independent effect of \({\tau }_{\dot{V}\mathrm{O}2}\) on CP could be observed. When exercise is initiated from an elevated baseline work rate, \({\tau }_{\dot{V}\mathrm{O}2}\) is greater than when compared with work initiated from a baseline of unloaded cycling [123,124,125,126]. Importantly, this slowing of the \(\dot{V}\)O2 kinetics appears to occur independently of any alterations in O2 availability [127,128,129].
Hence, we conducted two further studies that assessed the influence of exercise initiated from an elevated baseline work rate on \({\tau }_{\dot{V}\mathrm{O}2}\) and CP in the upright [130] and supine [117] positions. In both of these studies, \({\tau }_{\dot{V}\mathrm{O}2}\) was greater (i.e. \(\dot{V}\)O2 kinetics was slower) and CP was correspondingly reduced during work-to-work exercise compared with when exercise was initiated from a baseline of unloaded cycling [117, 130]. Crucially, indicators of O2 availability determined via NIRS were either improved [130] or unchanged [117] during work initiated from an elevated baseline, suggesting that the slowing of \(\dot{V}\)O2p kinetics brought about by this intervention was wholly independent of changes in microvascular O2 availability. Taken together, these findings therefore demonstrate an independent effect of \({\tau }_{\dot{V}\mathrm{O}2}\) on CP [130], and that this effect persisted even in situations where O2 delivery is substantially impaired [117].
The determining effect of \({\tau }_{\dot{V}\mathrm{O}2}\) on CP observed in healthy populations [64, 114, 117, 130] was later confirmed in a study that assessed the impact of priming exercise on \(\dot{V}\)O2 kinetics and CP in a population of individuals with type 1 diabetes mellitus [131]. In this population, priming exercise speeded \(\dot{V}\)O2 kinetics and increased CP during subsequent severe-intensity cycle exercise. Notably, these effects were accompanied by a concomitant speeding of muscle deoxygenation kinetics determined via NIRS [131]. As the muscle deoxygenation signal derived via NIRS represents the relative balance between O2 delivery and utilisation within the interrogated region, a relative speeding of muscle deoxygenation kinetics suggests that the effects of priming exercise on \({\tau }_{\dot{V}\mathrm{O}2}\) were predominantly due to an upregulation of otherwise impaired intracellular mechanisms of mitochondrial O2 utilisation, rather than O2 delivery [131]. Taken together, therefore, substantial recent evidence has accumulated to demonstrate that rates of intracellular O2 utilisation at the onset of exercise, encapsulated by \({\tau }_{\dot{V}\mathrm{O}2}\), can influence CP independently of factors related to mitochondrial O2 provision.
3 Interaction of Factors Determining CP
The studies of Goulding et al. [8, 64, 65, 114, 117, 130, 131] provide convincing evidence that \({\tau }_{\dot{V}\mathrm{O}2}\) is an independent determinant of CP. As reviewed above, there is also evidence for an independent determining role of convective and diffusive O2 delivery in influencing CP. That each of \({\tau }_{\dot{V}\mathrm{O}2}\), convective and diffusive O2 delivery has an independent role in determining CP is evinced by the fact that each can alter CP without a concomitant change in the other.
The proportion of CP explained by \({\tau }_{\dot{V}\mathrm{O}2}\) has been reported to be as high as 90% in a homogenous participant group where relative exercise intensity was precisely controlled (i.e., a tolerable duration of 6 min across subjects) [14]. Our own data have demonstrated R2 values of 0.64–0.90 for the relationship between CP and \({\tau }_{\dot{V}\mathrm{O}2}\) during upright exercise [60, 111, 127, 128]. Collation of these data across differing exercise intensity domains and populations, including hyperoxia conditions, yields an R2 = 0.60 (Fig. 3A; data from [131] previously unpublished), with a slope of ~ 0.03 W kg−1 s−1. However, this includes data from diseased populations (type 1 diabetes [131]) and hyperoxia [65], both of which might be expected to confound the analysis as the latter may distort the relationship between pulmonary and muscle \({\tau }_{\dot{V}\mathrm{O}2}\) and the former has a slope (0.01 W kg−1 s−1) significantly different to the healthy populations. Exclusion of diseased and hyperoxic data blunts the strength of the relationship between CP and \({\tau }_{\dot{V}\mathrm{O}2}\) (R2 = 0.43; Fig. 3B). However, the strength of this relationship increases markedly when only moderate-intensity exercise in healthy participants is considered (R2 = 0.79; Fig. 3C). The slope of the relationship between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP was preserved across this latter analysis, and taken together, CP appears to be well predicted from \({\tau }_{\dot{V}\mathrm{O}2}\) when the latter is precisely determined for a given relative exercise intensity, varying by ~ 0.03 W kg−1 per second change in \({\tau }_{\dot{V}\mathrm{O}2}\). However, and perhaps exemplified by the data from type 1 diabetes [131] and hyperoxia [65], when this relationship is expanded to cover the range of values for \({\tau }_{\dot{V}\mathrm{O}2}\) encountered across the animal kingdom (Fig. 3D), the relationship with CP appears curvilinear, but nevertheless preserved, suggesting a fundamental linkage of CP with muscular bioenergetics across species. Moreover, when the human-only data are considered and the speed of oxygen uptake kinetics expressed as a rate constant (i.e., 1/\({\tau }_{\dot{V}\mathrm{O}2}\)), the relationship with CP is linear (Fig. 3E). Accordingly, when the scope of human aerobic fitness is considered, the relationship between CP and \({\tau }_{\dot{V}\mathrm{O}2}\) can be considered to be hyperbolic, with previously published linear relationships [14, 104] being an artefact of participant homogeneity. By contrast, only one previous study has titrated the effect of oxygen delivery on CP [63]. Here, the reduction in CP with increasing altitude as a proxy for oxygen delivery was established, simulated by changes to FiO2. A non-linear (third-order polynomial) relationship was established with increases in altitude producing progressively larger reductions in CP. Critical power was reduced by 74 W with a 4000-m increase in altitude, though any such relationship will inevitably be impacted by the effect of reductions in FiO2 increasing \({\tau }_{\dot{V}\mathrm{O}2}\) (i.e. slowing \(\dot{V}\)O2 kinetics).

Panels A–C show the relationship between the fundamental phase time constant of pulmonary oxygen uptake kinetics (\({\tau }_{\dot{V}\mathrm{O}2}\)) and critical power normalized by body mass across a series of four experiments performed by Goulding et al. [65, 114, 130, 131]. Panel A displays all conditions from these studies in which \({\tau }_{\dot{V}\mathrm{O}2}\) was characterised with a high degree of confidence, including both moderate- and heavy-intensity exercise, normoxia and hyperoxia (fraction of inspired O2 = 0.5), and in patients with type 1 diabetes mellitus. Panel B displays the same relationship with removal of data points where \({\tau }_{\dot{V}\mathrm{O}2}\) was characterised in hyperoxic conditions and in type 1 diabetes (see “Sect. 2” for discussion). Panel C displays the relationship when only normoxic moderate-intensity exercise transitions in healthy participants are utilised. Note the increase in the R2 value as the conditions become more uniform with respect to exercise intensity, population and fraction of inspired O2. Panel D shows the relationship between \({\tau }_{\dot{V}\mathrm{O}2}\) and critical \(\dot{V}\)O2 across various human populations; elite athletes [157], young trained, active young, healthy elderly, and patients with chronic obstructive pulmonary disease (COPD) and other species where measurements of \({\tau }_{\dot{V}\mathrm{O}2}\) and critical power have both been conducted (i.e. the thoroughbred racehorse, rat, ghost crab and lungless salamander). The figure is derived from values reported in the literature of 28 papers published between 1982 and 2010; human populations were originally reported by Rossiter [104], with groups which were approximately matched for age, \(\dot{V}\)O2max and health status. Table S1 of the Electronic Supplementary Material should be consulted for details regarding derivation of critical \(\dot{V}\)O2 in different species. Panel E shows human-only data from panel D of critical power (CP) [mL kg−1 min−1] plotted as a function of 1/\({\tau }_{\dot{V}\mathrm{O}2}\) (i.e. the rate constant, k). There is a notable linear relationship across what can be regarded as the complete range of human fitness, indicating that the relationship between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP is hyperbolic, with previously published linear relationships likely being a function of participant homogeneity, and thus reflecting only a truncated portion of the hyperbolic relationship
Given the evidence reviewed herein, we therefore propose that each of mitochondrial O2 utilisation (encapsulated by the \({\tau }_{\dot{V}\mathrm{O}2}\) parameter), convective and diffusive O2 delivery exert independent effects on CP such that intracellular O2 utilisation and O2 transport interact to determine CP (Fig. 4). Exceptions to this include where pulmonary limitations (e.g. [34]) are dominant factors in limiting exercise tolerance to the extent that they dictate the shape of the power–duration relationship.

Schematic illustrating each of the factors that has been demonstrated to impact upon critical power. Convective and diffusive O2 delivery act in concert with muscle O2 utilisation to determine the degree of intracellular metabolic perturbation and fatigue induction incurred during the rest-to-exercise transition. The extent of such metabolic perturbations, in turn, determines whether an exercise bout can be met in a metabolic steady state within a given myocyte. Within a given individual, whether an extant power output is met in a whole-body steady state will depend on the muscle fibre-type composition of the individual, the muscle recruitment patterns employed during the task, and the extent of metabolic derangement and fatigue induction incurred in the recruited fibres during the rest-to-exercise transition. This figure was created with BioRender.com and was exported under a paid subscription. CaO2 arterial oxygen content, DO2 muscle diffusive capacity, PO2im intra-myocyte O2 pressure, PO2cap capillary O2 pressure, \(\dot{Q}\), cardiac output
The precise mechanisms underpinning such an interaction have not been fully elucidated; however, a starting point is to consider the inexorable loss of intracellular homeostasis, and thus unsustainable rise in O2 deficit, during exercise above, but not below, CP. This is accompanied by a mirror-like association between peripheral fatigue [107, 132] and the loss of exercise efficiency [109, 133, 134] that occurs during exercise above CP [135]. Of the factors that accumulate as a result of the O2 deficit, [Pi] is a prime candidate for the common denominator between fatigue and efficiency owing to its central role in muscle fatigue and task failure [136]. A recent in silico study by Korzeniewski and Rossiter [10] tested the hypothesis that accumulation of [Pi] during the transition from rest to work could explain both the loss of intracellular homeostasis during supra-CP exercise and the fatigue-related termination of exercise. Using a validated model of the human bioenergetic system, Korzeniewski and Rossiter [10] defined a “critical” (i.e. threshold) [Pi] above which further [Pi] accumulation drove an increase in the requirements for ATP turnover (i.e. an increased ATP cost of muscle contraction) and a “peak” (i.e. limiting) [Pi] at which exercise would cease. The additional ATP turnover driven by [Pi] accumulation resulted in a self-propagating positive feedback loop where additional ATP turnover resulted in increased [Pi], which caused fatigue and additional ATP turnover, until the pre-defined peak [Pi] (and accompanying muscle \(\dot{V}\)O2max) was achieved. By contrast, when [Pi] accumulated below or only marginally above critical [Pi], this positive feedback loop stabilised such that [Pi] did not attain peak values and muscle oxygen uptake attained a steady state. Based on these findings, we therefore recently proposed a model whereby muscle O2 consumption kinetics determine CP by dictating the magnitude of O2 deficit (and thus [Pi], amongst other factors) accumulated during a given exercise transition [8]. Slow \(\dot{V}\)O2 kinetics begets large intracellular perturbations whereas fast \(\dot{V}\)O2 kinetics engenders smaller intracellular perturbations for a given metabolic rate at exercise onset [102, 104, 110, 137]. Accordingly, more rapid \(\dot{V}\)O2 kinetics will enable a higher exercise intensity before a critical value of [Pi] is breached, thereby increasing critical power, all else being equal. Importantly, simulating alterations in PO2im within the computer model of Korzeniewski and Rossiter [10] resulted in the changes in \({\tau }_{\dot{V}\mathrm{O}2}\) and CP predicted by the evidence reviewed in each of the previous sections [10].
Alongside other O2 deficit-related factors, that breaching a critical [Pi] results in an inexorable cascade of increasing [Pi], fatigue and ATP turnover is also consistent with the evidence reviewed herein whereby convective and diffusive O2 delivery has a determining effect on CP. O2 delivery is known to regulate the concentrations of phosphate metabolites at a given metabolic rate, such that when intracellular PO2 is higher, the intracellular perturbations incurred for [Pi], [PCr] and [ADP] are reduced, whereas the reverse is true when intracellular PO2 is lower [49, 50, 54, 138]. From these observations, it follows that the aforementioned effects of convective and diffusive O2 delivery and intracellular O2 utilisation on CP stem from their impact upon the intracellular metabolic state, or more specifically, the rate of ATP turnover at which a critical threshold for [Pi] (which is itself a proxy for a collection of intracellular metabolites reflecting the intracellular state of fatigue) is attained. Hence, faster \(\dot{V}\)O2 kinetics, as well as increased O2 delivery, exert their effects on CP via reducing the intracellular metabolic perturbations required to sustain a given rate of ATP turnover, thus enabling a higher power output to be achieved before CP is reached.
4 Integration of Mechanisms: Whole Body
This model of CP being an emergent property of the metabolic derangements established at the onset of exercise may provide an explanation for the metabolic bases of CP at the level of a single fibre; however, it does not pretend to be a complete explanation of CP at the integrative whole-body level. This is despite the in silico approach of Korzeniewski and Rossiter [10] being “chimeric”, in that it is built using the data of whole-body and whole-muscle responses of \(\dot{V}\)O2, [PCr], [Pi] and pH and reflecting a variety of muscle fibre types, averaged into a single response. In practice, the exercise transition is undertaken by muscle fibres across the spectrum of function, with differing underlying oxidative phosphorylation activities, each-step activation intensities, convective and diffusive O2 supply, and fatigue characteristics [139,140,141]. Additionally, the location of a given fibre with respect to the skin surface has implications for the relative O2 delivery [45, 118,119,120, 142]. Nevertheless, findings at the whole muscle level are congruent with the notion that metabolic inertia at the onset of exercise determines CP via its effect on the accumulation of Pi and other O2 deficit-related metabolites that are implicated in the fatigue process. Type I fibres possess faster \(\dot{V}\)O2 kinetics, better metabolic control, and maintain greater values for capillary and interstitial PO2 at rest and during contractions [139, 140, 143,144,145,146,147,148,149,150]. Hence, as detailed earlier, in human biopsy studies, the proportion of type I fibres and indices of muscle fibre capillarisation have been shown to be closely associated with CP [31, 86].
Moreover, given that type I fibres maintain greater values for capillary and interstitial PO2 at rest and during contractions (presumably due to enhanced capillarisation) [139, 140, 143,144,145,146,147,148,149,150], these data are consistent with the present proposal that \({\tau }_{\dot{V}\mathrm{O}2}\), convective and diffusive O2 delivery each exert independent interactive determining effects on CP. However, the eventual external outcome of interest from all of these processes, i.e. CP, will also be a function of factors such as (relative) exercising muscle mass, the local musculoskeletal lever system dynamics and co-ordination, the extent of localised fatigue within working muscle groups and motor-unit recruitment. Indeed, that our data demonstrate a significant relationship between \({\tau }_{\dot{V}\mathrm{O}2}\) and CP expressed in W kg−1, but not W (data not shown), speaks to the role of exercising muscle mass in the eventual determination of CP. The individual muscles of the quadriceps muscle group have been shown to produce divergent patterns of [PCr] depletion and [Pi] accumulation within distinct muscle regions (72-cm3 voxels) during fatiguing (incremental) exercise [151]. Exercising muscle mass may therefore also play a role in the extent of such muscle metabolite heterogeneity, and thus the degree of metabolic perturbation within distinct muscle regions, which in turn contributes towards setting CP.
Morgan and colleagues [152, 153] provided insight into how muscle recruitment patterns may act to determine CP. During repeated intermittent isometric contractions, the ingestion of acetaminophen led to a smaller reduction in torque across 60 MVCs when compared with a placebo [153]. This was associated with a greater preservation of muscle activation with acetaminophen as assessed via electromyography. Subsequently, it was shown that during upright cycle ergometry, acute acetaminophen ingestion increased CP and preserved muscle activity throughout the duration of exercise when compared with a placebo [152]. These findings suggest blunting neuromuscular fatigue development and preserving muscle activation enhances CP, and thus demonstrates the importance of motor unit recruitment profiles.
The interaction between muscle recruitment patterns and muscle O2 delivery in determining CP is perhaps most strikingly illustrated by the recent study of Hammer et al. [78], discussed previously (see Sect. 2.1). These authors showed that, following muscle reperfusion, both muscular activity and force production returned to levels not different from those observed under free-flowing conditions [78]. Hence, muscle occlusion constrained muscular recruitment and thus critical force; however, once muscle perfusion was restored to pre-occlusion conditions, both muscle recruitment and force-generating capacity were restored. These findings illustrate that CP represents an intricate balance between muscle O2 supply, muscle recruitment patterns and peripheral fatigue development.
To summarise, CP is sensitive to muscle fibre type composition because it is a parameter of aerobic function. Hence, the oxidative characteristics inherent within type I fibres, such as rapid \(\dot{V}\)O2 kinetics, greater rates of blood flow, and higher capillary and interstitial PO2 values, allow the attainment of high rates of ATP utilisation with minimal derangement of the intracellular metabolic milieu. Therefore, all else being equal, individuals with a relatively greater proportion of type I skeletal muscle fibres will tend to possess greater CP values when compared with individuals of equivalent training status with a greater proportion of type II fibres. Moreover, animal data indicate that CP appears to be a critical threshold for the recruitment of high-order motor units containing a high fraction of type II fibres [25]. Hence, individuals with more type I fibres will attain a relatively greater fraction of their \(\dot{V}\)O2 max before reaching the threshold for progressive recruitment of type II fibres, i.e. CP (as seen in highly trained humans, [111, 154]). Interventions that increase motor unit recruitment are also conducive to high CP values, as a greater number of motor units/muscle fibres performing a given task will lessen the metabolic strain on each individual fibre. Hence, when muscular recruitment is increased, each fibre is able to maintain intramuscular metabolite accumulation below its critical threshold for a wider range of ATP utilisation rates, thus enabling a greater CP, as suggested for the effects of priming by Burnley et al. [155]. Therefore, although there is clearly a role for convective and diffusive O2 delivery and intracellular O2 utilisation in determining CP, understanding the physiology of CP at the level of integrative physiology is only possible via consideration of how these factors interact with muscle fibre-type composition and recruitment patterns.
5 Conclusions
Critical power separates the heavy and severe exercise-intensity domains wherein qualitatively divergent physiological responses are observed, such that CP represents the threshold intensity above which a metabolic steady state cannot be attained during exercise. Hence, CP is fundamental to the understanding of human endurance performance and the causes of exercise limitation in populations where exercise tolerance is impaired. Over the past 15 years or so, evidence has emerged that CP also represents a key threshold for a variety of aspects of physiological system behaviour, such as muscle fibre recruitment, blood flow and vascular control, as well as muscle fatigue. Accordingly, a wide range of evidence has emerged, spanning each step of the oxygen transport pathway, that CP is a fundamental parameter of aerobic function. It has been demonstrated that alterations in delivery of O2 to the exercising muscles, via both convection and diffusion, impact upon CP. The rates of O2 utilisation during exercise, particularly during the transition from rest to work, also play a key role in determining CP by governing the degree of matching between the rates of ATP utilisation and production. These factors each interact with one another, and via this interaction determine the degree of intracellular metabolic disturbance required to sustain a given power output. How each of these factors interacts to determine CP at the whole-body level will be dependent upon the muscle fibre-type composition and their recruitment patterns during exercise.
References
Burnley M, Jones AM. Oxygen uptake kinetics as a determinant of sports performance. Eur J Sport Sci. 2007;7:63–79.
Joyner MJ, Coyle EF. Endurance exercise performance: the physiology of champions. J Physiol. 2008;586:35–44.
Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346:793–801.
Myers J, Kaykha A, George S, Abella J, Zaheer N, Lear S, et al. Fitness versus physical activity patterns in predicting mortality in men. Am J Med. 2004;117:912–8.
Black MI, Jones AM, Blackwell JR, Bailey SJ, Wylie LJ, McDonagh STJ, et al. Muscle metabolic and neuromuscular determinants of fatigue during cycling in different exercise intensity domains. J Appl Physiol. 2017;122:446–59.
Poole DC, Ward SA, Gardner GW, Whipp BJ. Metabolic and respiratory profile of the upper limit for prolonged exercise in man. Ergonomics. 1988;31:1265–79.
Wasserman K, Hansen JE, Sietsema K, Sue DY, MD WWS, Whipp B, et al. Principles of exercise testing and interpretation: including pathophysiology and clinical applications. Philadelphia, Pennsylvania: Wolters Kluwer Health; 2015.
Goulding RP, Rossiter HB, Marwood S, Ferguson C. Bioenergetic mechanisms linking VO2 kinetics and exercise tolerance. Exerc Sport Sci Rev. 2021. https://journals.lww.com/acsm-essr/Abstract/9000/Bioenergetic_Mechanisms_Linking_V_Combining_Dot.99696.aspx. Accessed 20 Jul 2021.
Gaesser GA, Poole DC. The slow component of oxygen uptake kinetics in humans. Exerc Sport Sci Rev. 1996;24:35–71.
Korzeniewski B, Rossiter HB. Exceeding a “critical” muscle Pi: implications for [Formula: see text] and metabolite slow components, muscle fatigue and the power-duration relationship. Eur J Appl Physiol. 2020;120(7):1609–19.
Jones AM, Vanhatalo A, Burnley M, Morton RH, Poole DC. Critical power: implications for determination of VO2max and exercise tolerance. Med Sci Sports Exerc. 2010;42:1876–90.
Poole DC, Burnley M, Vanhatalo A, Rossiter HB, Jones AM. Critical power: an important fatigue threshold in exercise physiology. Med Sci Sports Exerc. 2016;48:2320–34.
Black MI, Durant J, Jones AM, Vanhatalo A. Critical power derived from a 3-min all-out test predicts 16.1-km road time-trial performance. Eur J Sport Sci. 2014;14:217–23.
Murgatroyd SR, Ferguson C, Ward SA, Whipp BJ, Rossiter HB. Pulmonary O2 uptake kinetics as a determinant of high-intensity exercise tolerance in humans. J Appl Physiol. 2011;110:1598–606.
Jones AM, Wilkerson DP, DiMenna F, Fulford J, Poole DC. Muscle metabolic responses to exercise above and below the “critical power” assessed using 31P-MRS. Am J Physiol Regul Integr Comp Physiol. 2008;294:R585–93.
Hill DW. The critical power concept. Sports Med. 1993;16:237–54.
Muniz-Pumares D, Karsten B, Triska C, Glaister M. Methodological approaches and related challenges associated with the determination of critical power and W. J Strength Cond Res. 2018;33:584–96.
MattioniMaturana F, Fontana FY, Pogliaghi S, Passfield L, Murias JM. Critical power: How different protocols and models affect its determination. J Sci Med Sport. 2017;21:742–7.
Jones AM, Burnley M, Black MI, Poole DC, Vanhatalo A. The maximal metabolic steady state: redefining the “gold standard.” Physiol Rep. 2019;7: e14098.
Monod H, Scherrer J. The work capacity of a synergic muscular group. Ergonomics. 1965;8:329–38.
Moritani T, Nagata A, deVries HA, Muro M. Critical power as a measure of physical work capacity and anaerobic threshold. Ergonomics. 1981;24:339–50.
Full RJ. Locomotion without lungs: energetics and performance of a lungless salamander. Am J Physiol. 1986;251:R775–80.
Full RJ, Herreid CF. Aerobic response to exercise of the fastest land crab. Am J Physiol. 1983;244:R530–6.
Lauderdale MA, Hinchcliff KW. Hyperbolic relationship between time-to-fatigue and workload. Equine Vet J Suppl. 1999;30:586–90.
Copp SW, Hirai DM, Musch TI, Poole DC. Critical speed in the rat: implications for hindlimb muscle blood flow distribution and fibre recruitment. J Physiol. 2010;588:5077–87.
Billat VL, Mouisel E, Roblot N, Melki J. Inter- and intrastrain variation in mouse critical running speed. J Appl Physiol Bethesda Md. 1985;2005(98):1258–63.
Burnley M. Estimation of critical torque using intermittent isometric maximal voluntary contractions of the quadriceps in humans. J Appl Physiol Bethesda Md. 1985;2009(106):975–83.
Smyth B, Muniz-Pumares D. Calculation of critical speed from raw training data in recreational marathon runners. Med Sci Sports Exerc. 2020;52:2637–45.
Smith CG, Jones AM. The relationship between critical velocity, maximal lactate steady-state velocity and lactate turnpoint velocity in runners. Eur J Appl Physiol. 2001;85:19–26.
Wakayoshi K, Ikuta K, Yoshida T, Udo M, Moritani T, Mutoh Y, et al. Determination and validity of critical velocity as an index of swimming performance in the competitive swimmer. Eur J Appl Physiol. 1992;64:153–7.
Vanhatalo A, Black MI, DiMenna FJ, Blackwell JR, Schmidt JF, Thompson C, et al. The mechanistic bases of the power-time relationship: muscle metabolic responses and relationships to muscle fibre type. J Physiol. 2016;594:4407–23.
Overend TJ, Cunningham DA, Paterson DH, Smith WD. Physiological responses of young and elderly men to prolonged exercise at critical power. Eur J Appl Physiol. 1992;64:187–93.
Mezzani A, Corrà U, Giordano A, Colombo S, Psaroudaki M, Giannuzzi P. Upper intensity limit for prolonged aerobic exercise in chronic heart failure. Med Sci Sports Exerc. 2010;42:633–9.
Neder JA, Jones PW, Nery LE, Whipp BJ. Determinants of the exercise endurance capacity in patients with chronic obstructive pulmonary disease: the power-duration relationship. Am J Respir Crit Care Med. 2000;162:497–504.
Neder JA, Jones PW, Nery LE, Whipp BJ. The effect of age on the power/duration relationship and the intensity-domain limits in sedentary men. Eur J Appl Physiol. 2000;82:326–32.
Barker AR, Bond B, Toman C, Williams CA, Armstrong N. Critical power in adolescents: physiological bases and assessment using all-out exercise. Eur J Appl Physiol. 2012;112:1359–70.
Vanhatalo A, Fulford J, DiMenna FJ, Jones AM. Influence of hyperoxia on muscle metabolic responses and the power-duration relationship during severe-intensity exercise in humans: a 31P magnetic resonance spectroscopy study. Exp Physiol. 2010;95:528–40.
Puente-Maestu L, SantaCruz A, Vargas T, Martínez-Abad Y, Whipp BJ. Effects of training on the tolerance to high-intensity exercise in patients with severe COPD. Respir Int Rev Thorac Dis. 2003;70:367–70.
van der Vaart H, Murgatroyd SR, Rossiter HB, Chen C, Casaburi R, Porszasz J. Selecting constant work rates for endurance testing in COPD: the role of the power-duration relationship. COPD. 2014;11:267–76.
Malaguti C, Nery LE, Dal Corso S, De Fuccio MB, Lerario MC, Cendon S, et al. Alternative strategies for exercise critical power estimation in patients with COPD. Eur J Appl Physiol. 2006;96:59–65.
Malaguti C, Dal Corso S, Colucci E, Stuchi T, Pulcheri R, Nery LE. Critical power for the upper limb in patients with chronic obstructive pulmonary disease: a pilot study. Respir Physiol Neurobiol. 2019;270: 103280.
Porszasz J, Rambod M, van der Vaart H, Rossiter HB, Ma S, Kiledjian R, et al. Sinusoidal high-intensity exercise does not elicit ventilatory limitation in chronic obstructive pulmonary disease. Exp Physiol. 2013;98:1102–14.
Dempsey JA, La Gerche A, Hull JH. Is the healthy respiratory system built just right, overbuilt, or underbuilt to meet the demands imposed by exercise? J Appl Physiol Bethesda Md. 1985;2020(129):1235–56.
Koga S, Poole DC, Ferreira LF, Whipp BJ, Kondo N, Saitoh T, et al. Spatial heterogeneity of quadriceps muscle deoxygenation kinetics during cycle exercise. J Appl Physiol. 2007;103:2049–56.
Koga S, Rossiter HB, Heinonen I, Musch TI, Poole DC. Dynamic heterogeneity of exercising muscle blood flow and O2 utilization. Med Sci Sports Exerc. 2014;46:860–76.
Heinonen I, Koga S, Kalliokoski KK, Musch TI, Poole DC. Heterogeneity of muscle blood flow and metabolism: influence of exercise, aging, and disease states. Exerc Sport Sci Rev. 2015;43:117–24.
MacDonald MJ, Tarnopolsky MA, Hughson RL. Effect of hyperoxia and hypoxia on leg blood flow and pulmonary and leg oxygen uptake at the onset of kicking exercise. Can J Physiol Pharmacol. 2000;78:67–74.
Welch HG, Bonde-Petersen F, Graham T, Klausen K, Secher N. Effects of hyperoxia on leg blood flow and metabolism during exercise. J Appl Physiol. 1977;42:385–90.
Hogan MC, Richardson RS, Haseler LJ. Human muscle performance and PCr hydrolysis with varied inspired oxygen fractions: a 31P-MRS study. J Appl Physiol Bethesda Md. 1985;1999(86):1367–73.
Haseler LJ, Kindig CA, Richardson RS, Hogan MC. The role of oxygen in determining phosphocreatine onset kinetics in exercising humans. J Physiol. 2004;558:985–92.
Cardinale DA, Larsen FJ, Jensen-Urstad M, Rullman E, Søndergaard H, Morales-Alamo D, et al. Muscle mass and inspired oxygen influence oxygen extraction at maximal exercise: role of mitochondrial oxygen affinity. Acta Physiol Oxf Engl. 2019;225: e13110.
Knight DR, Schaffartzik W, Poole DC, Hogan MC, Bebout DE, Wagner PD. Effects of hyperoxia on maximal leg O2 supply and utilization in men. J Appl Physiol Bethesda Md. 1985;1993(75):2586–94.
Calbet JA. Oxygen tension and content in the regulation of limb blood flow. Acta Physiol Scand. 2000;168:465–72.
Hogan MC, Arthur PG, Bebout DE, Hochachka PW, Wagner PD. Role of O2 in regulating tissue respiration in dog muscle working in situ. J Appl Physiol Bethesda Md. 1985;1992(73):728–36.
Richardson RS, Noyszewski EA, Leigh JS, Wagner PD. Lactate efflux from exercising human skeletal muscle: role of intracellular PO2. J Appl Physiol Bethesda Md. 1985;1998(85):627–34.
Dekerle J, Mucci P, Carter H. Influence of moderate hypoxia on tolerance to high-intensity exercise. Eur J Appl Physiol. 2012;112:327–35.
Williams JH, Powers SK, Stuart MK. Hemoglobin desaturation in highly trained athletes during heavy exercise. Med Sci Sports Exerc. 1986;18:168–73.
Powers SK, Lawler J, Dempsey JA, Dodd S, Landry G. Effects of incomplete pulmonary gas exchange on VO2 max. J Appl Physiol Bethesda Md. 1985;1989(66):2491–5.
Gore CJ, Hahn AG, Scroop GC, Watson DB, Norton KI, Wood RJ, et al. Increased arterial desaturation in trained cyclists during maximal exercise at 580 m altitude. J Appl Physiol Bethesda Md. 1985;1996(80):2204–10.
Simpson LP, Jones AM, Skiba PF, Vanhatalo A, Wilkerson D. Influence of hypoxia on the power-duration relationship during high-intensity exercise. Int J Sports Med. 2015;36:113–9.
Valli G, Cogo A, Passino C, Bonardi D, Morici G, Fasano V, et al. Exercise intolerance at high altitude (5050 m): critical power and W’. Respir Physiol Neurobiol. 2011;177:333–41.
La Monica MB, Fukuda DH, Starling-Smith TM, Wang R, Hoffman JR, Stout JR. Effects of normobaric hypoxia on upper body critical power and anaerobic working capacity. Respir Physiol Neurobiol. 2017;249:1–6.
Townsend NE, Nichols DS, Skiba PF, Racinais S, Périard JD. Prediction of critical power and W’ in hypoxia: application to work-balance modelling. Front Physiol. 2017;8:180.
Goulding RP, Roche DM, Marwood S. Hyperoxia speeds pulmonary oxygen uptake kinetics and increases critical power during supine cycling. Exp Physiol. 2019;104:1061–73.
Goulding RP, Roche DM, Marwood S. Effect of hyperoxia on critical power and VO2 kinetics during upright cycling. Med Sci Sports Exerc. 2019;52:1041–9.
Hoelting BD, Scheuermann BW, Barstow TJ. Effect of contraction frequency on leg blood flow during knee extension exercise in humans. J Appl Physiol Bethesda Md. 1985;2001(91):671–9.
Lutjemeier BJ, Miura A, Scheuermann BW, Koga S, Townsend DK, Barstow TJ. Muscle contraction-blood flow interactions during upright knee extension exercise in humans. J Appl Physiol Bethesda Md. 1985;2005(98):1575–83.
Sadamoto T, Bonde-Petersen F, Suzuki Y. Skeletal muscle tension, flow, pressure, and EMG during sustained isometric contractions in humans. Eur J Appl Physiol. 1983;51:395–408.
Robergs RA, Icenogle MV, Hudson TL, Greene ER. Temporal inhomogeneity in brachial artery blood flow during forearm exercise. Med Sci Sports Exerc. 1997;29:1021–7.
Barcroft H, Dornhorst AC. The blood flow through the human calf during rhythmic exercise. J Physiol. 1949;109:402–11.
Folkow B, Gaskell P, Waaler BA. Blood flow through limb muscles during heavy rhythmic exercise. Acta Physiol Scand. 1970;80:61–72.
Walløe L, Wesche J. Time course and magnitude of blood flow changes in the human quadriceps muscles during and following rhythmic exercise. J Physiol. 1988;405:257–73.
Broxterman AdCJ, Wilcox SL, Schlup SJ, Craig JC, Barstow TJ. Influence of duty cycle on the power-duration relationship: observations and potential mechanisms. Respir Physiol Neurobiol. 2014;192:102–11.
Broxterman AdCJ, Craig JC, Wilcox SL, Schlup SJ, Barstow TJ. Influence of blood flow occlusion on muscle oxygenation characteristics and the parameters of the power-duration relationship. J Appl Physiol Bethesda Md. 1985;2015(118):880–9.
Broxterman AdCJ, Craig JC, Wilcox SL, Barstow TJ. The effect of resting blood flow occlusion on exercise tolerance and W’. Am J Physiol Regul Integr Comp Physiol. 2015;309:R684–91.
Hamaoka T, Iwane H, Shimomitsu T, Katsumura T, Murase N, Nishio S, et al. Noninvasive measures of oxidative metabolism on working human muscles by near-infrared spectroscopy. J Appl Physiol Bethesda Md. 1985;1996(81):1410–7.
Hamaoka T, Osada T, Murase N, Sako T, Higuchi H, Kurosawa Y, et al. Quantitative evaluation of oxygenation and metabolism in the human skeletal muscle. Opt Rev. 2003;10:493–7.
Hammer SM, Alexander AM, Didier KD, Barstow TJ. Influence of blood flow occlusion on muscular recruitment and fatigue during maximal-effort small muscle-mass exercise. J Physiol. 2020;598:4293–306.
Hammer SM, Alexander AM, Didier KD, Huckaby LM, Barstow TJ. Limb blood flow and muscle oxygenation responses during handgrip exercise above vs. below critical force. Microvasc Res. 2020;131:104002.
Hammer SM, Hammond ST, Parr SK, Alexander AM, Turpin V-RG, White ZJ, et al. Influence of muscular contraction on vascular conductance during exercise above versus below critical power. Respir Physiol Neurobiol. 2021;293:103718.
Richardson RS, Poole DC, Knight DR, Kurdak SS, Hogan MC, Grassi B, et al. High muscle blood flow in man: is maximal O2 extraction compromised? J Appl Physiol Bethesda Md. 1985;1993(75):1911–6.
Federspiel WJ, Popel AS. A theoretical analysis of the effect of the particulate nature of blood on oxygen release in capillaries. Microvasc Res. 1986;32:164–89.
Groebe K, Thews G. Calculated intra- and extracellular PO2 gradients in heavily working red muscle. Am J Physiol. 1990;259:H84-92.
Richardson RS, Grassi B, Gavin TP, Haseler LJ, Tagore K, Roca J, et al. Evidence of O2 supply-dependent VO2 max in the exercise-trained human quadriceps. J Appl Physiol Bethesda Md. 1985;1999(86):1048–53.
Richardson RS, Leigh JS, Wagner PD, Noyszewski EA. Cellular PO2 as a determinant of maximal mitochondrial O(2) consumption in trained human skeletal muscle. J Appl Physiol Bethesda Md. 1985;1999(87):325–31.
Mitchell EA, Martin NRW, Bailey SJ, Ferguson RA. Critical power is positively related to skeletal muscle capillarity and type I muscle fibers in endurance trained individuals. J Appl Physiol. 2018;125(3):737–45.
Ansdell P, Brownstein CG, Škarabot J, Hicks KM, Howatson G, Thomas K, et al. Sex differences in fatigability and recovery relative to the intensity–duration relationship. J Physiol. 2019;597:5577–95.
Ansdell P, Škarabot J, Atkinson E, Corden S, Tygart A, Hicks KM, et al. Sex differences in fatigability following exercise normalised to the power–duration relationship. J Physiol. 2020;598:5717–37.
Roepstorff C, Steffensen CH, Madsen M, Stallknecht B, Kanstrup I-L, Richter EA, et al. Gender differences in substrate utilization during submaximal exercise in endurance-trained subjects. Am J Physiol Endocrinol Metab. 2002;282:E435–47.
Simoneau JA, Bouchard C. Human variation in skeletal muscle fiber-type proportion and enzyme activities. Am J Physiol. 1989;257:E567–72.
Staron RS, Hagerman FC, Hikida RS, Murray TF, Hostler DP, Crill MT, et al. Fiber type composition of the vastus lateralis muscle of young men and women. J Histochem Cytochem. 2000;48:623–9.
Andersen P, Adams RP, Sjøgaard G, Thorboe A, Saltin B. Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol Bethesda Md. 1985;1985(59):1647–53.
Cleland SM, Murias JM, Kowalchuk JM, Paterson DH. Effects of prior heavy-intensity exercise on oxygen uptake and muscle deoxygenation kinetics of a subsequent heavy-intensity cycling and knee-extension exercise. Appl Physiol Nutr Metab. 2012;37:138–48.
Krustrup P, Söderlund K, Mohr M, González-Alonso J, Bangsbo J. Recruitment of fibre types and quadriceps muscle portions during repeated, intense knee-extensor exercise in humans. Pflugers Arch. 2004;449:56–65.
Koga S, Poole DC, Shiojiri T, Kondo N, Fukuba Y, Miura A, et al. Comparison of oxygen uptake kinetics during knee extension and cycle exercise. Am J Physiol Regul Integr Comp Physiol. 2005;288:R212–20.
Koga S, Okushima D, Poole DC, Rossiter HB, Kondo N, Barstow TJ. Unaltered Vo2 kinetics despite greater muscle oxygenation during heavy-intensity two-legged knee extension versus cycle exercise in humans. Am J Physiol Regul Integr Comp Physiol. 2019;317:R203–13.
Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD. Myoglobin O2 desaturation during exercise: evidence of limited O2 transport. J Clin Invest. 1995;96:1916–26.
Volianitis S, Secher NH. Cardiovascular control during whole body exercise. J Appl Physiol Bethesda Md. 1985;2016(121):376–90.
Poole DC, Copp SW, Hirai DM, Musch TI. Dynamics of muscle microcirculatory and blood-myocyte O(2) flux during contractions. Acta Physiol Oxf Engl. 2011;202:293–310.
Colburn TD, Weber RE, Hageman KS, Caldwell JT, Schulze KM, Ade CJ, et al. Vascular ATP-sensitive K+ channels support maximal aerobic capacity and critical speed via convective and diffusive O2 transport. J Physiol. 2020;598:4843–58.
Poole DC, Jones AM. Oxygen uptake kinetics. Compr Physiol. 2012;2:933–96.
Grassi B, Porcelli S, Salvadego D, Zoladz JA. Slow VO2 kinetics during moderate-intensity exercise as markers of lower metabolic stability and lower exercise tolerance. Eur J Appl Physiol. 2011;111:345–55.
Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol Bethesda Md. 1985;1996(80):988–98.
Rossiter HB. Exercise: kinetic considerations for gas exchange. Compr Physiol. 2011;1:203–44.
Whipp BJ. Rate constant for the kinetics of oxygen uptake during light exercise. J Appl Physiol. 1971;30:261–3.
Whipp BJ, Ward SA, Lamarra N, Davis JA, Wasserman K. Parameters of ventilatory and gas exchange dynamics during exercise. J Appl Physiol. 1982;52:1506–13.
Keir DA, Copithorne DB, Hodgson MD, Pogliaghi S, Rice CL, Kowalchuk JM. The slow component of pulmonary O2 uptake accompanies peripheral muscle fatigue during high-intensity exercise. J Appl Physiol. 2016;121:493–502.
Temesi J, Mattioni Maturana F, Peyrard A, Piucco T, Murias JM, Millet GY. The relationship between oxygen uptake kinetics and neuromuscular fatigue in high-intensity cycling exercise. Eur J Appl Physiol. 2017;117:969–78.
Cannon DT, White AC, Andriano MF, Kolkhorst FW, Rossiter HB. Skeletal muscle fatigue precedes the slow component of oxygen uptake kinetics during exercise in humans. J Physiol. 2011;589:727–39.
Korzeniewski B, Rossiter HB. Factors determining training-induced changes in VO2max, critical power and VO2 on-kinetics in skeletal muscle. J Appl Physiol. 2021;130:498–507.
Jones AM. The physiology of the world record holder for the women’s marathon. Int J Sports Sci Coach. 2006;1:101–16.
Koppo K, Bouckaert J, Jones AM. Effects of training status and exercise intensity on phase II VO2 kinetics. Med Sci Sports Exerc. 2004;36:225–32.
Murias JM, Kowalchuk JM, Paterson DH. Speeding of VO2 kinetics in response to endurance-training in older and young women. Eur J Appl Physiol. 2011;111:235–43.
Goulding RP, Roche DM, Marwood S. Prior exercise speeds pulmonary oxygen uptake kinetics and increases critical power during supine but not upright cycling. Exp Physiol. 2017;102:1158–76.
Jones AM, Berger NJA, Wilkerson DP, Roberts CL. Effects of “priming” exercise on pulmonary O2 uptake and muscle deoxygenation kinetics during heavy-intensity cycle exercise in the supine and upright positions. J Appl Physiol Bethesda Md. 1985;2006(101):1432–41.
Koga S, Shiojiri T, Shibasaki M, Kondo N, Fukuba Y, Barstow TJ. Kinetics of oxygen uptake during supine and upright heavy exercise. J Appl Physiol Bethesda Md. 1985;1999(87):253–60.
Goulding RP, Roche DM, Marwood S. “Work-to-work” exercise slows pulmonary oxygen uptake kinetics, decreases critical power, and increases W’ during supine cycling. Physiol Rep. 2018;6: e13916.
Goulding RP, Marwood S, Okushima D, Poole DC, Barstow TJ, Lei T-H, et al. Effect of priming exercise and body position on pulmonary oxygen uptake and muscle deoxygenation kinetics during cycle exercise. J Appl Physiol. 2020;129(4):810–22.
Goulding RP, Okushima D, Marwood S, Poole DC, Barstow TJ, Lei T-H, et al. Impact of supine exercise on muscle deoxygenation kinetics heterogeneity: mechanistic insights into slow pulmonary oxygen uptake dynamics. J Appl Physiol Bethesda Md. 1985. 2020;129(3):535–46.
Goulding RP, Okushima D, Fukuoka Y, Marwood S, Kondo N, Poole DC, et al. Impact of supine versus upright exercise on muscle deoxygenation heterogeneity during ramp incremental cycling is site specific. Eur J Appl Physiol. 2021;121(5):1283–96.
Gurd BJ, Peters SJ, Heigenhauser GJF, LeBlanc PJ, Doherty TJ, Paterson DH, et al. Prior heavy exercise elevates pyruvate dehydrogenase activity and muscle oxygenation and speeds O2 uptake kinetics during moderate exercise in older adults. Am J Physiol Regul Integr Comp Physiol. 2009;297:R877–84.
Hernández A, McDonald JR, Lai N, Gladden LB. A prior bout of contractions speeds VO2 and blood flow on-kinetics and reduces the VO2 slow-component amplitude in canine skeletal muscle contracting in situ. J Appl Physiol Bethesda Md. 1985;2010(108):1169–76.
Brittain CJ, Rossiter HB, Kowalchuk JM, Whipp BJ. Effect of prior metabolic rate on the kinetics of oxygen uptake during moderate-intensity exercise. Eur J Appl Physiol. 2001;86:125–34.
di Prampero PE, Mahler PB, Giezendanner D, Cerretelli P. Effects of priming exercise on VO2 kinetics and O2 deficit at the onset of stepping and cycling. J Appl Physiol Bethesda Md. 1985;1989(66):2023–31.
DiMenna FJ, Bailey SJ, Vanhatalo A, Chidnok W, Jones AM. Elevated baseline VO2 per se does not slow O2 uptake kinetics during work-to-work exercise transitions. J Appl Physiol Bethesda Md. 1985;2010(109):1148–54.
MacPhee SL, Shoemaker JK, Paterson DH, Kowalchuk JM. Kinetics of O2 uptake, leg blood flow, and muscle deoxygenation are slowed in the upper compared with lower region of the moderate-intensity exercise domain. J Appl Physiol Bethesda Md. 1985;2005(99):1822–34.
Bowen TS, Murgatroyd SR, Cannon DT, Cuff TJ, Lainey AF, Marjerrison AD, et al. A raised metabolic rate slows pulmonary O2 uptake kinetics on transition to moderate-intensity exercise in humans independently of work rate. Exp Physiol. 2011;96:1049–61.
DiMenna FJ, Wilkerson DP, Burnley M, Jones AM. Influence of priming exercise on pulmonary O2 uptake kinetics during transitions to high-intensity exercise from an elevated baseline. J Appl Physiol Bethesda Md. 1985;2008(105):538–46.
Wüst RCI, McDonald JR, Sun Y, Ferguson BS, Rogatzki MJ, Spires J, et al. Slowed muscle oxygen uptake kinetics with raised metabolism are not dependent on blood flow or recruitment dynamics. J Physiol. 2014;592:1857–71.
Goulding RP, Roche DM, Marwood S. Elevated baseline work rate slows pulmonary oxygen uptake kinetics and decreases critical power during upright cycle exercise. Physiol Rep. 2018;6(14):e13802.
Goulding RP, Roche DM, Scott SN, Koga S, Weston PJ, Marwood S. Limitations to exercise tolerance in type 1 diabetes: the role of pulmonary oxygen uptake kinetics and priming exercise. J Appl Physiol. 2020;128:1299–309.
Burnley M, Vanhatalo A, Jones AM. Distinct profiles of neuromuscular fatigue during muscle contractions below and above the critical torque in humans. J Appl Physiol Bethesda Md. 1985;2012(113):215–23.
Zoladz JA, Gladden LB, Hogan MC, Nieckarz Z, Grassi B. Progressive recruitment of muscle fibers is not necessary for the slow component of VO2 kinetics. J Appl Physiol Bethesda Md. 1985;2008(105):575–80.
Vanhatalo A, Poole DC, DiMenna FJ, Bailey SJ, Jones AM. Muscle fiber recruitment and the slow component of O2 uptake: constant work rate vs. all-out sprint exercise. Am J Physiol Regul Integr Comp Physiol. 2011;300:R700–7.
Grassi B, Rossiter HB, Zoladz JA. Skeletal muscle fatigue and decreased efficiency: two sides of the same coin? Exerc Sport Sci Rev. 2015;43:75–83.
Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332.
Rossiter HB, Ward SA, Kowalchuk JM, Howe FA, Griffiths JR, Whipp BJ. Effects of prior exercise on oxygen uptake and phosphocreatine kinetics during high-intensity knee-extension exercise in humans. J Physiol. 2001;537:291–303.
Hogan MC, Nioka S, Brechue WF, Chance B. A 31P-NMR study of tissue respiration in working dog muscle during reduced O2 delivery conditions. J Appl Physiol Bethesda Md. 1985;1992(73):1662–70.
Behnke BJ, McDonough P, Padilla DJ, Musch TI, Poole DC. Oxygen exchange profile in rat muscles of contrasting fibre types. J Physiol. 2003;549:597–605.
McDonough P, Behnke BJ, Padilla DJ, Musch TI, Poole DC. Control of microvascular oxygen pressures in rat muscles comprised of different fibre types. J Physiol. 2005;563:903–13.
Ferreira LF, McDonough P, Behnke BJ, Musch TI, Poole DC. Blood flow and O2 extraction as a function of O2 uptake in muscles composed of different fiber types. Respir Physiol Neurobiol. 2006;153:237–49.
Koga S, Okushima D, Barstow TJ, Rossiter HB, Kondo N, Poole DC. Near-infrared spectroscopy of superficial and deep rectus femoris reveals markedly different exercise response to superficial vastus lateralis. Physiol Rep. 2017;5: e13402.
Wüst RCI, van der Laarse WJ, Rossiter HB. On-off asymmetries in oxygen consumption kinetics of single Xenopus laevis skeletal muscle fibres suggest higher-order control. J Physiol. 2013;591:731–44.
Crow MT, Kushmerick MJ. Chemical energetics of slow- and fast-twitch muscles of the mouse. J Gen Physiol. 1982;79:147–66.
Jackman MR, Willis WT. Characteristics of mitochondria isolated from type I and type IIb skeletal muscle. Am J Physiol. 1996;270:C673–8.
Willis WT, Jackman MR. Mitochondrial function during heavy exercise. Med Sci Sports Exerc. 1994;26:1347–53.
Barstow TJ, Jones AM, Nguyen PH, Casaburi R. Influence of muscle fiber type and pedal frequency on oxygen uptake kinetics of heavy exercise. J Appl Physiol Bethesda Md. 1985;1996(81):1642–50.
Pringle JSM, Doust JH, Carter H, Tolfrey K, Campbell IT, Sakkas GK, et al. Oxygen uptake kinetics during moderate, heavy and severe intensity “submaximal” exercise in humans: the influence of muscle fibre type and capillarisation. Eur J Appl Physiol. 2003;89:289–300.
Colburn TD, Hirai DM, Craig JC, Ferguson SK, Weber RE, Schulze KM, et al. Transcapillary PO2 gradients in contracting muscles across the fibre type and oxidative continuum. J Physiol. 2020;598:3187–202.
Hirai DM, Craig JC, Colburn TD, Eshima H, Kano Y, Sexton WL, et al. Skeletal muscle microvascular and interstitial PO2 from rest to contractions. J Physiol. 2017;596:869–83.
Cannon DT, Howe FA, Whipp BJ, Ward SA, McIntyre DJ, Ladroue C, et al. Muscle metabolism and activation heterogeneity by combined 31P chemical shift and T2 imaging, and pulmonary O2 uptake during incremental knee-extensor exercise. J Appl Physiol. 2013;115:839–49.
Morgan PT, Vanhatalo A, Bowtell JL, Jones AM, Bailey SJ. Acetaminophen ingestion improves muscle activation and performance during a 3-min all-out cycling test. Appl Physiol Nutr Metab Physiol Appl Nutr Metab. 2019;44:434–42.
Morgan PT, Bowtell JL, Vanhatalo A, Jones AM, Bailey SJ. Acute acetaminophen ingestion improves performance and muscle activation during maximal intermittent knee extensor exercise. Eur J Appl Physiol. 2018;118:595–605.
Jones AM, Poole DC. Physiological demands of endurance exercise. In: Olympic textbook of science in sport. Amsterdam: Wiley; 2008. p. 43–55.
Burnley M, Doust JH, Ball D, Jones AM. Effects of prior heavy exercise on VO(2) kinetics during heavy exercise are related to changes in muscle activity. J Appl Physiol Bethesda Md. 1985;2002(93):167–74.
Wasserman K, Van Kessel AL, Burton GG. Interaction of physiological mechanisms during exercise. J Appl Physiol. 1967;22:71–85.
Jones AM, Kirby BS, Clark IE, Rice HM, Fulkerson E, Wylie LJ, et al. Physiological demands of running at 2-hour marathon race pace. J Appl Physiol. 2021;130:369–79.
Gaesser GA, Wilson LA. Effects of continuous and interval training on the parameters of the power-endurance time relationship for high-intensity exercise. Int J Sports Med. 1988;9:417–21.
Poole DC, Ward SA, Whipp BJ. The effects of training on the metabolic and respiratory profile of high-intensity cycle ergometer exercise. Eur J Appl Physiol. 1990;59:421–9.
Jenkins DG, Quigley BM. Endurance training enhances critical power. Med Sci Sports Exerc. 1992;24:1283–9.
Hill DW, Smith JC, Leuschel JL, Chasteen SD, Miller SA. Effect of pedal cadence on parameters of the hyperbolic power-time relationship. Int J Sports Med. 1995;16:82–7.
Serres I, Varray A, Vallet G, Micallef JP, Préfaut C. Improved skeletal muscle performance after individualized exercise training in patients with chronic obstructive pulmonary disease. J Cardiopulm Rehabil Prev. 1997;17:232–8.
Barker T, Poole DC, Noble ML, Barstow TJ. Human critical power–oxygen uptake relationship at different pedalling frequencies. Exp Physiol. 2006;91:621–32.
Vanhatalo A, Doust JH, Burnley M. A 3-min all-out cycling test is sensitive to a change in critical power. Med Sci Sports Exerc. 2008;40:1693–9.
Miura A, Shiragiku C, Hirotoshi Y, Kitano A, Endo MY, Barstow TJ, et al. The effect of prior heavy exercise on the parameters of the power-duration curve for cycle ergometry. Appl Physiol Nutr Metab. 2009;34:1001–7.
Corn SD, Barstow TJ. Effects of oral N-acetylcysteine on fatigue, critical power, and W’ in exercising humans. Respir Physiol Neurobiol. 2011;178:261–8.
Mueller SM, Aguayo D, Lunardi F, Ruoss S, Boutellier U, Frese S, et al. High-load resistance exercise with superimposed vibration and vascular occlusion increases critical power, capillaries and lean mass in endurance-trained men. Eur J Appl Physiol. 2014;114:123–33.
Broxterman RM, Ade CJ, Barker T, Barstow TJ. Influence of pedal cadence on the respiratory compensation point and its relation to critical power. Respir Physiol Neurobiol. 2015;208:1–7.
Black MI, Jones AM, Bailey SJ, Vanhatalo A. Self-pacing increases critical power and improves performance during severe-intensity exercise. Appl Physiol Nutr Metab. 2015;40:662–70.
Deb SK, Gough LA, Sparks SA, McNaughton LR. Determinants of curvature constant (W’) of the power duration relationship under normoxia and hypoxia: the effect of pre-exercise alkalosis. Eur J Appl Physiol. 2017;117:901–12.
Clark IE, Vanhatalo A, Bailey SJ, Wylie LJ, Kirby BS, Wilkins BW, et al. Effects of two hours of heavy-intensity exercise on the power-duration relationship. Med Sci Sports Exerc. 2018;50:1658–68.
Clark IE, Vanhatalo A, Thompson C, Joseph C, Black MI, Blackwell JR, et al. Dynamics of the power-duration relationship during prolonged endurance exercise and influence of carbohydrate ingestion. J Appl Physiol. 2019;127:726–36.
Clark IE, Vanhatalo A, Thompson C, Wylie LJ, Bailey SJ, Kirby BS, et al. Changes in the power-duration relationship following prolonged exercise: estimation using conventional and all-out protocols and relationship with muscle glycogen. Am J Physiol Regul Integr Comp Physiol. 2019;317:R59-67.
Waldron M, Patterson SD, Jeffries O. Oral taurine improves critical power and severe-intensity exercise tolerance. Amino Acids. 2019;51:1433–41.
Karabiyik H, Eser MC, Guler O, Yasli BC, Ertetik G, Sisman A, et al. The effects of 15 or 30 s SIT in normobaric hypoxia on aerobic, anaerobic performance and critical power. Int J Environ Res Public Health. 2021;18:3976.
Collins J, Leach O, Dorff A, Linde J, Kofoed J, Sherman M, et al. Critical power and work-prime account for variability in endurance training adaptations not captured by Vo2max. J Appl Physiol Bethesda Md. 1985;2022(133):986–1000.
Byrd MT, Switalla JR, Eastman JE, Wallace BJ, Clasey JL, Bergstrom HC. Contributions of body-composition characteristics to critical power and anaerobic work capacity. Int J Sports Physiol Perform. 2018;13:189–93.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Richie Goulding was funded by a European Foundation for the Study of Diabetes Boehringer Ingelheim European Research Programme grant during the completion of this work. No specific sources of funding were used to assist in the preparation of this article.
Conflicts of interests/competing interests
Richie P. Goulding and Simon Marwood have no conflicts of interest that are directly relevant to the content of this review.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
RPG conceived the work and wrote the first draft. Both authors conducted the literature search, revised the manuscript critically for important intellectual content and approved the final version to be submitted. Both authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Goulding, R.P., Marwood, S. Interaction of Factors Determining Critical Power. Sports Med 53, 595–613 (2023). https://doi.org/10.1007/s40279-022-01805-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40279-022-01805-w