
Abstract
Background
Spinal muscular atrophy is a rare neuromuscular disorder with a spectrum of severity related to age at onset and the number of SMN2 gene copies. Infantile-onset (≤ 6 months of age) is the most severe spinal muscular atrophy and is the leading monogenetic cause of infant mortality; patients with later-onset (> 6 months of age) spinal muscular atrophy can survive into adulthood. Nusinersen is a new treatment for spinal muscular atrophy.
Objective
The objective of this study was to evaluate the cost effectiveness of nusinersen for the treatment of patients with infantile-onset spinal muscular atrophy and later-onset spinal muscular atrophy in Sweden.
Methods
One Markov cohort health-state transition model was developed for each population. The infantile-onset and later-onset models were based on the efficacy results from the ENDEAR phase III trial and the CHERISH phase III trial, respectively. The cost effectiveness of nusinersen in both models was compared with standard of care in Sweden.
Results
For a time horizon of 40 years in the infantile-onset model and 80 years in the later-onset model, treatment with nusinersen resulted in 3.86 and 9.54 patient incremental quality-adjusted life-years and 0.02 and 2.39 caregiver incremental quality-adjusted life-years and an incremental cost of 21.9 and 38.0 million SEK (Swedish krona), respectively. These results translated into incremental cost-effectiveness ratios (including caregiver quality-adjusted life-years) of 5.64 million SEK (€551,300) and 3.19 million SEK (€311,800) per quality-adjusted life-year gained in the infantile-onset model and later-onset model, respectively.
Conclusions
Treatment with nusinersen resulted in overall survival and quality-adjusted life-year benefits but with incremental costs above 21 million SEK (€2 million) [mainly associated with maintenance treatment with nusinersen over a patient’s lifespan]. Nusinersen was not cost effective when using a willingness-to-pay threshold of 2 million SEK (€195,600), which has been considered in a recent discussion by the Dental and Pharmaceutical Benefits Agency as a reasonable threshold for rare disease. Nonetheless, nusinersen gained reimbursement in Sweden in 2017 for paediatric patients (below 18 years old) with spinal muscular atrophy type I–IIIa.
Similar content being viewed by others

1 Introduction
Spinal muscular atrophy (SMA) is a rare, progressive, autosomal recessive neuromuscular disease caused by decreased levels of functional survival of motor neuron (SMN) protein, leading to a loss of motor function and to respiratory failure [1]. Presentation of SMA adheres to a spectrum based on age at illness onset and highest motor function achieved: onset in utero (type 0), infantile-onset (most likely to develop type I), later-onset (most likely to develop type II or type III) and adult-onset (type IV) SMA [2]. Symptom onset and severity of SMA generally correlate with SMN2 gene copy number [3]. Although rare, SMA is a leading genetic cause of infant mortality. If untreated, patients with in-utero SMA onset do not survive beyond the first months of life, [4] and patients with infantile-onset SMA, the most common form (by incidence), never achieve the ability to sit without support, with a life span of <2 years without respiratory support and nutritional intervention [5]. Patients with later-onset SMA range from those who can sit independently but never stand or walk to those who are able to stand and walk but fall frequently and have trouble walking upstairs and later lose ambulation [6, 7]. An infant with normal development will learn to maintain a sitting position without support and will learn to walk at a median age of 6.5 months and 12.8 months, respectively [8]. Adult onset is the least severe form of SMA, and its occurrence is rare; patients are ambulatory and have a normal life expectancy, but experience mild-to-moderate muscle weakness [9].
The reported incidence for types I–III combined is similar among studies, ranging from 8.5 to 10.3 per 100,000 live births [10,11,12,13,14]. In Western Sweden, the incidences for SMA type I, type II, and type III were 3.6, 2.1 and 2.8 per 100,000 live births, respectively [10]. Because of the increased death risk for the more severe types, the population prevalence is higher for the less severe types. A study in Northern England estimated population-based point prevalences per 100,000 of 0.10, 0.57 and 0.64 for SMA type I, type II, and type III, respectively [15].
Standard of care (SoC) recommendations for SMA have been published by the International Standard of Care Committee for SMA [2; updated in 2018: 16, 17] and the French Muscular Dystrophy Association in conjunction with Coordination des Centres de Référence Neuromusculaires [18]. Disease management in the guidelines before 2018 targeted only respiratory and nutritional symptoms and orthopaedic function decline. The updated 2018 guidelines include nusinersen as a new treatment for SMA [16, 17].
Despite improvements in disease management care, developmental milestones are rarely even partially achieved in patients with infantile-onset SMA, [19] and pulmonary disease secondary to neuromuscular weakness is a major cause of morbidity and mortality [2]. Almost all patients with infantile-onset SMA eventually need essential nutritional support via a feeding tube [20]. Patients with later-onset SMA are at risk of significant scoliosis, which is managed with surgical spine correction, as well as pulmonary disease [2, 21].
Nusinersen is an antisense oligonucleotide designed to treat SMA caused by mutations or deletions in the survival of motor neuron 1 (SMN1) gene on chromosome 5q. The SMN1 gene lies on the fifth chromosome in the region labelled “q”. Therefore, SMA types I–IV are often referred to as “5q SMA.” These mutations lead to deficiency of the SMN protein. Nusinersen modifies pre-messenger RNA splicing of SMN2 to promote increased production of full-length SMN protein [22]. Nusinersen is delivered intrathecally by lumbar puncture once every 4 months (following four loading doses) directly into the cerebrospinal fluid. Clinical trial programmes have evaluated the efficacy and safety of nusinersen across a wide spectrum of patients with SMA: infantile-onset (ENDEAR, EMBRACE and CS3A [22,23,24]), later-onset (CHERISH, EMBRACE, CS2, CS12, CS1 and CS10 [25,26,27,28,29,30]) and presymptomatic (NURTURE [31]). Patients who previously participated in the nusinersen clinical trial programme [22,23,24,25,26,27,28,29,30] could move to an open-label extension study (SHINE) [32].
The primary efficacy endpoints in the nusinersen infantile-onset clinical trial (ENDEAR) were the Hammersmith Infant Neurological Examination Sect. 2 [33] and event-free survival (death or permanent ventilation), with the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) [34], overall survival, permanent ventilation and electrophysiological measures as secondary endpoints. Compared with patients receiving the sham procedure plus SoC (SoC arm hereafter), patients who received nusinersen plus SoC in the ENDEAR trial showed statistically significant and clinically meaningful improvement in motor milestones, as well as sustained and clinically meaningful improvements in event-free survival, overall survival (OS), motor function and motor neuron health [22]. The primary endpoint in the nusinersen later-onset clinical trial (CHERISH) was the Hammersmith Functional Motor Scale-Expanded (HFMSE) [35, 36], with the World Health Organization (WHO) motor milestones [37] and the revised Upper Limb Module test [38] as secondary endpoints. Compared to patients in the control group, patients who received nusinersen in the CHERISH trial showed statistically significant improvement in HFMSE scores [26]. Nusinersen has been approved by both the US Food and Drug Administration and the European Medicines Agency for the treatment of SMA [39, 40].
Economic modelling in rare diseases can be challenging owing to limitations in the available input parameter data and structural assumptions [41]. This is also the case in SMA where there are no existing published models and there are data gaps for long-term natural history, cost and utility weight data. Our objective was to develop two separate economic models to evaluate the cost effectiveness of nusinersen in treating patients with infantile- and later-onset SMA compared to SoC. The models were developed from the societal perspective in Sweden; results from the payer perspective were reported in sensitivity analyses.
2 Methods
Cost-utility models for infantile- and later-onset SMA were based on Markov health-state structures. Development of the model structures was informed by both clinical and health economic experts. One UK clinical expert, chosen because of the clinician’s involvement in clinical trials of nusinersen, provided initial input, while seven UK paediatric neuromuscular disease specialists were chosen to attend an advisory board because of their experience in treating patients with SMA. Health economic input was sought via face-to-face interviews with two UK health economists, who were chosen for their expertise in knowing the requirements for economic submissions to health technology assessment bodies throughout the world. Cost and health-state utility assumptions were validated by one Swedish clinical expert. Data inputs were informed by clinical and economic systematic literature reviews and reviewed by UK and Swedish clinical experts. The economic systematic literature review [Table S1 and Fig. S1 of the Electronic Supplementary Material (ESM)] identified limited resource use and cost data and did not identify existing published economic models or utility weights. Therefore, de novo models for infantile-onset and later-onset SMA were developed and additional studies were performed to derive country-specific cost estimates and health-state utilities.
2.1 Model Structures
2.1.1 Infantile-Onset Spinal Muscular Atrophy
The infantile-onset SMA model structure comprised ten health states (Fig. 1a). A response-based section of the model structure defined patients by their change in CHOP INTEND score from baseline (worsened, stabilisation of baseline function and improvement health states). Scores on the CHOP INTEND range from 0 to 64, with higher scores indicating better motor function [34]. Improvement is defined as at least a 4-point increase from baseline (same response definition used in the ENDEAR trial), and worsened is defined as at least a 4-point decrease from baseline (stabilisation of baseline function is defined as a change from baseline between − 3 and 3 points). Although the literature does not report a defined minimal clinically important difference for the CHOP INTEND score change, results from the ENDEAR trial support the view that a 4-point change increase is rarely observed in natural history, with a significantly higher percentage of infants in the nusinersen group achieving response compared with the SoC arm (71% vs. 3%) [22]. A natural history study by Finkel et al. [42] reported a CHOP INTEND score decline of 1.27 [95% confidence interval (CI) − 2.33 to − 0.21] points per annum for patients with infantile-onset SMA.

Infantile-onset (a) and later-onset (b) spinal muscular atrophy model structures. In the infantile-onset model, improvement is defined in the CHOP INTEND scale as: at least a 4-point increase from baseline. Worsened is defined as at least a 4-point decrease from baseline. Hammersmith Infant Neurological Examination scores are used to determine patients achieving later-onset milestones. In the later-onset model, an increase of less than 3 points in the Hammersmith Functional Motor Scale-Expanded (HFMSE) score is considered mild; an increase greater than or equal to 3 points is considered moderate. A decrease in score of 1 or more points is considered as worsening. World Health Organization measures are used to determine patients achieving type III motor milestones
A second section divided patients into motor milestone-defined health states (according to Sect. 2 of the Hammersmith Infant Neurological Examination scale. The scores ranged from 0 to 26 [through the achievement of motor milestones], with higher scores indicating better motor function [33]) not usually observed in patients with infantile-onset SMA (sits without support, stands with assistance, walks with assistance and stands/walks unaided health states) and a health state (loss of later-onset motor function) that captured patients who had previously been able to sit without support but then lost that ability. Patients entered the model in the stabilisation of baseline function health state. The model structure included health states uncharacteristic of patients with infantile-onset SMA because interim data from ENDEAR and 26-month data from CS3A in symptomatic patients with infantile-onset SMA showed that patients treated with nusinersen achieved motor milestones typical of later types of SMA.
A patient remained in the response-based section of the model until the patient achieved a milestone characteristic of later-onset SMA (sits without support). Once a patient has achieved a later-onset milestone (as observed in the ENDEAR trial for patients in the nusinersen arm), the patient could either continue to achieve higher motor milestones or could lose the ability to sit without support and move to the loss of later-onset motor function health state. This latter health state was included to explore scenarios where the quality of life and costs associated with a patient who was not able to sit without support differ between patients that at some point had achieved the sitting without support milestone (or higher) and patients that never achieved that milestone.
2.1.2 Later-Onset Spinal Muscular Atrophy
The model structure for later-onset SMA incorporated changes from baseline HFMSE scores (scores range from 0 to 66, with higher scores indicating better motor function [35]) to determine the membership in the response-based health states (worse than baseline, stabilisation of baseline function, mild increase HFMSE score and moderate increase in HFMSE score) and achievement of motor milestones characteristic of patients with SMA type III, as assessed by the WHO motor milestones criteria (stands/walks with assistance, stands unaided and walks unaided) (Fig. 1b) [37]. The WHO motor milestones are commonly used in clinical practice. The later-onset SMA model structure also included one health state that captured patients who had previously been able to stand or walk with assistance but then lost that ability (lost ambulation with/without assistance health state) and the dead health state.
In the model, an increase from baseline in the HFMSE score of less than 3 points is considered mild, a score increase of 3 or more points is considered moderate and a score decrease of 1 or more points is considered worsening (stabilisation of baseline function is defined as no change from baseline). The definition of mild and moderate improvement was included to differentiate patients who had score improvements not normally observed in natural history (i.e. those with moderate improvement). A study by Mercuri et al. [43] reported that an increase of greater than two points was unlikely in patients with later-onset SMA. In addition, patients and caregivers considered a 1-point increase or even stabilisation as meaningful. [44, 45]
A patient remained in the response-based section of the model until the patient achieved a motor milestone uncharacteristic of patients with SMA type II (stands/walks with assistance). Once a patient has achieved the stands/walks with assistance milestone, the patient could either continue to achieve higher motor milestones (characteristic of SMA type III) or move to the loss of ambulation with/without assistance health state. This latter health state was included to explore scenarios where the quality of life and costs associated with a patient who was not able to stand/walk with assistance differ between patients that at some point had achieved the stand/walk with assistance milestone (or higher) and patients who never achieved that milestone.
2.2 Patient Characteristics
The model populations were patients with infantile-onset and later-onset SMA. The patient cohort for the infantile-onset model had the same starting age as that of patients who received their first dose in the ENDEAR trial (Table 1). The starting age of the later-onset model cohort matched the mean age at screening observed in the CHERISH trial (Table 2).
2.3 Analytical Perspective and Time Horizon
The models were developed from the societal perspective in Sweden (direct medical costs [primary and secondary care], patients’ reimbursed transportation costs and informal care costs; Dental and Pharmaceutical Benefits Agency [TLV] guidelines [46]). The payer perspective also was considered in sensitivity analyses.
A time horizon of 40 years was used in the infantile-onset model because data from clinical trials suggested that treatment with nusinersen had a significant effect on survival [the ENDEAR trial reported an OS hazard ratio of 0.37 (95% CI 0.32–0.89; p = 0.004) and the phase II dose-escalation trial indicated a significant differentiation in age at death or permanent ventilation compared with a natural history case series (log-rank test; p = 0.0014)], [22, 23] which potentially could be associated with the achievement of motor milestones similar to those achieved by patients in the later-onset model. The later-onset model used a default time horizon of 80 years, informed by data from Zerres et al. [47] that were used to guide OS after the trial follow-up.
In both models, the model cycles during the trial follow-up were aligned with the motor function assessment time points (ENDEAR: days 1, 64, 183, 302 and 394; CHERISH: days 1, 92, 169, 274, 365 and 456). After the trial follow-up, the models used a cycle length of 4 months to match the 4-month maintenance dosing interval specified by the European Medicines Agency [40]. Costs other than drug costs after the trial follow-up were based on cost-per-year estimates. A half-cycle correction was applied for both models.
Both costs and outcomes were discounted at 3.0% (Pharmaceutical Benefits Board [46]; TLV guidelines). Model outcomes included incremental cost per quality-adjusted life-year (QALY) gained (base-case scenario included QALYs gained for both patients and caregivers) and OS.
2.4 Intervention and Comparators
The costs and outcomes for nusinersen and SoC were compared with those for SoC alone, as nusinersen is currently the only approved treatment for SMA. Safety and efficacy of alternative treatments are being evaluated in phase I–III studies [48].
2.5 Disease Progression and Death: Trial Follow-up Period
Probabilities of transitioning to health states other than death within the trial follow-up period were calculated from the individual patient’s motor function score and motor milestone achievement at the different assessment points in the ENDEAR and CHERISH trials (Tables S9–S30 of the ESM).
For predicting OS for patients with infantile-onset SMA, parametric survival functions were fitted to observed OS data in the ENDEAR trial in accordance with TLV guidelines and based on the Decision Support Unit guidance for fitting and selecting survival functions from the UK National Institute for Health and Care Excellence [49]. Alternative assumptions regarding the continuation of the treatment effect after the trial follow-up and treatment discontinuation were explored.
The clinical and biological plausibility of OS prediction was explored by comparison with external long-term survival data [7, 50]. Potential limitations associated with the use of these data include possible differences in SoC, patient characteristics and improvements in respiratory management. Fitting parametric survival functions to the ENDEAR data without the use of external data was not considered appropriate for this analysis (OS predictions did not appear clinically plausible).
For patients with later-onset SMA, the model assumed that, within the trial follow-up period, nusinersen had no effect on improving the mortality rates for these patients, as no patients in either treatment arm in the CHERISH trial died. Survival after the trial follow-up was guided by long-term OS data presented by Zerres et al. [47] (see Sect. 2.7.2).
2.6 Disease Progression to Health States Other Than Dead After the Trial Follow-Up
2.6.1 Infantile-Onset Spinal Muscular Atrophy
The model assigned a CHOP INTEND score to each health state based on the mean CHOP INTEND score of the patients in each health state at the end of the ENDEAR trial follow-up. Because patients in the ENDEAR trial did not achieve the walks with assistance and the stand/walks unaided milestones, data from the last follow-up in the CS3A trial were used to assign the CHOP INTEND score to those health states. To estimate the probability of transitioning to the next best or next worst health state, the model used the rate of CHOP INTEND score change. For patients in the nusinersen arm, the score increased 0.251 (95% CI 0.19–0.32) points per week, while the score for patients in the SoC arm decreased 0.364 (95% CI 0.27–0.46) points per week, which is consistent with natural history [42]. Therefore, the base-case analysis assumed that patients in the nusinersen arm continue to improve after the trial follow-up and that patients in the SoC arm transition to worse health states after the trial follow-up according to the corresponding rate of the CHOP INTEND score change. A scenario analysis assumed that a proportion of patients in the nusinersen arm reached an improvement plateau, with some of those patients progressing as in the SoC arm.
2.6.2 Later-Onset Spinal Muscular Atrophy
Similar to the infantile-onset model, the transition probabilities to health states other than death were based on the rate of motor function score change. The model assigned a HFMSE score to each health state based on the mean HFMSE score of the patients in each health state at the end of the CHERISH trial follow-up. The HFMSE score of the walking unaided health state was determined using 35-month long-term data from the CS2/12 trial. Patients in the nusinersen arm in the CHERISH trial had a change from baseline of 3.9 (95% CI 3.00–4.90) points at 15 months, while the mean HFMSE score change from baseline in the SoC arm was − 1.0 (95% CI − 2.50 to 0.50) points at 15 months [26]. The HFMSE score change at the interim data cut was included as a scenario analysis (Table S7 of the ESM). Hence, after the trial follow-up in the base-case analysis, patients in the nusinersen arm moved to the next best health state while patients in the SoC arm transitioned to the next worse health state.
2.7 External Long-Term Overall Survival Data After the Trial Follow-up
2.7.1 Infantile-Onset Spinal Muscular Atrophy
External long-term data identified in the literature have shown large differences in survival between countries [5, 7, 50, 51]. This could be attributed to heterogenous patient populations and the type of care provided (i.e. invasive or not invasive respiratory support). Gregoretti et al. [50] reported OS for three groups of patients receiving different types of respiratory support. Survival at 24 months for the no respiratory support group, the non-invasive ventilation group and the invasive ventilation group was 1.3% (95% CI, 0.1–6), 67.7% (95% CI 46.7–82) and 95% (95% CI 81.8–8.8), respectively. Survival at 24 months in other studies [5, 7, 51] ranged from 32 to 74%.
Feedback from clinical experts has shown that invasive respiration is generally not used in Sweden, resulting in an expected survival of less than 3 years (usually 1 year; TLV, 2017 [52]); therefore, survival data for respiratory care without invasive ventilation were used in the base case. For this scenario, the method of least squares [53] was used to fit parametric functions to the Kaplan–Meier data presented by Gregoretti et al. [50] for a group of patients that did not receive respiratory support.
After the trial follow-up, patients in the SoC arm were assumed to have the same hazard rate as that from the function fitted to data in Gregoretti et al. [50] (Fig. 2). Different scenarios were used for the nusinersen arm, including assuming an indefinite treatment effect, no residual treatment effect and a gradual decline. The base-case analysis assumed that the treatment effect continues indefinitely.

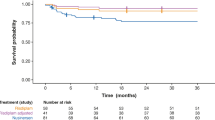
Base-case overall survival prediction: infantile-onset spinal muscular atrophy (SMA) a Flexible spline-based Weibull function with two knots was fitted to ENDEAR trial data during the trial follow-up. b After the trial follow-up (i.e. after 13 months), the hazard from the log-normal function fitted to the Gregoretti et al. [50] Kaplan–Meier (KM) data were applied to the standard of care (SoC) function. c After the trial follow-up, the treatment effect from the trial was applied indefinitely; patients achieving motor milestones consistent with later-onset SMA were applied a risk for death in between the risk for death of patients with infantile-onset SMA and the risk for death of patients with later-onset SMA (adjustment factor, 0.9). The risk for death of patients with later-onset SMA was determined from the flexible spline-based Weibull function with two knots fitted to the overall survival (OS) KM for patients later-onset SMA (type II) presented by Zerres et al. [47]. The model assumes the same risk for death as in the real world care arm for patients who discontinue treatment
The base case also included an assumption that patients achieving motor milestones consistent with later-onset SMA have a lower risk of death than patients who never achieve these milestones. Zerres et al. [47] reported OS for patients with SMA type II. To estimate the risk of death for patients with later-onset SMA, a flexible spline-based Weibull (2 knots) function was fitted to the Kaplan–Meier from Zerres et al. [47]. The risk of death for patients achieving later-onset milestones was assumed to be between the risk of death of a patient with infantile-onset SMA and the risk of death of a patient with later-onset SMA. The model used an adjustment factor between 0 and 1, where 0 resulted in the same risk of death for patients with infantile-onset SMA and 1 resulted in the same risk of death for patients with later-onset SMA. The base case used an adjustment factor of 0.9 based on clinical opinion (Fig. 2). Time-dependent transition probabilities to death were calculated from the parametric survival functions.
2.7.2 Later-Onset Spinal Muscular Atrophy
No deaths occurred in the CHERISH trial. The later-onset SMA model used long-term OS data for patients with later-onset SMA (type II) presented by Zerres et al. [47]. From the studies reporting long-term survival for patients with SMA type II [5, 47, 54, 55], only Zerres et al. [47] and Farrar et al. [5] reported survival estimates beyond 10 years. However, the model used the data from Zerres et al. [47] as the sample in that study was more robust than the sample in Farrar et al. [5] (240 patients in Germany and Poland compared with 30 patients in Australia). Parametric models were selected based on Akaike information criterion results, Bayesian information criterion results, integrated Brier score through bootstrap cross validation, the visual fit of the function to the Kaplan–Meier data and the assessment of the clinical plausibility of the extrapolated portion of the survival curve. [56] The flexible spline-based Weibull function with two knots was selected as the base case because it provided the best visual fit.
After the trial follow-up, patients on the SoC arm continued to die according to OS data from Zerres et al. [47]. Scenarios explored for the nusinersen arm included assumptions that there was no treatment effect (base-case analysis), the treatment effect continued (using a placeholder hazard ratio) and a tapering of the treatment effect over time.
The base-case analysis assumed that patients achieving advanced motor milestones characteristic of later-onset SMA (i.e. stands unaided, walks unaided) had a lower risk for death than patients in other health states. Patients with SMA and advanced motor milestone characteristics have a life expectancy that is not reported to be significantly less that the general population [5]. It was assumed in the base-case analysis that the risk of death for patients with advanced motor milestone characteristics is between that of patients with later-onset SMA (type II) and that of the general population (adjustment factor of 0.5 was applied) (Fig. 3). Time-dependent transition probabilities to death were calculated from the parametric survival functions.

Base-case overall survival prediction: later-onset spinal muscular atrophy (SMA). Flexible spline-based Weibull model with two knots fitted to the Zerres et al. [47] data; no treatment effect after the trial follow up; lower risk for death (adjustment factor of 0.5) to patients in health states consistent with advance motor milestones of later-onset SMA (type III: stand unaided and walks unaided); same risk for death as in the standard of care (SoC) arm for patients who discontinue treatment
2.8 Resource Use and Costs
Resource use data for the models were collected from interviews with two Swedish clinical experts, as the economic literature review (Table S1 and Fig. S1 of the ESM) presented resource use and costs from only a German study [57] and a Spanish study [58]. In accordance with Swedish guidelines for health economic evaluations, the societal perspective was applied, including caregiver production loss for one caregiver in accordance with clinical expert input. Production loss was estimated based on the monthly salary and the assumption (according to the Swedish clinical experts) that a caregiver of a patient with infantile-onset SMA would give up work completely, while a caregiver of a patient with type II SMA would give up work for 6 months during the first year and for about 3 months the following year. Country-specific unit costs were mainly collected from hospital price lists. When available, prices were gathered for 2016 and inflated to the year 2018 level according to the Swedish Consumer Price Index. The resulting per-patient annual resource use and total costs of infantile- and later-onset SMA in Sweden for the first and the following years are summarised in Tables S2–4 of the ESM. These per-patient annual costs were then assigned to each health state in the model (Tables 1, 2: health-state costs). If the health state was associated with milestones characteristic of SMA type I, the model assigned the infantile-onset SMA costs; for health states associated with milestones characteristic of SMA type II, the model assigned the later-onset SMA costs; and for health states associated with milestones characteristic of SMA type III, the model assigned 25% of the later-onset SMA costs (assumption validated by Swedish clinical experts).
Nusinersen is administered via lumbar puncture with four loading doses and a maintenance dose every 4 months thereafter [40]. With a list price of 800,757 SEK (Swedish krona) per vial (€78,300Footnote 1), [59] the first year of treatment with nusinersen was calculated to be 4.8 million SEK (€469,500) per patient and 2.4 million SEK (€234,800) per year per patient in subsequent years. Because nusinersen is a new medicine, it was assumed to be administered in inpatient care, which was verified by Swedish experts. Hence, the procedure cost for lumbar puncture was retrieved from the hospital price list for the Southern Health Care Region in Sweden, which amounted to 3308 SEK (code TAB00; Södra Regionvårdsnämnden [60]).
The cost for production loss should be added to administration costs, according to TLV guidelines. [46] However, it was assumed that production loss as a result of drug administration is already taken into account as caregiver production loss.
The base-case analysis assumed that patients treated with nusinersen spend less time in hospital and require less ventilatory support than patients in the SoC arm. Results from the ENDEAR trial supported this assumption, as the overall proportion of time spent hospitalised was significantly lower in the nusinersen arm than the SoC arm (least-square mean treatment difference − 0.093; 95% CI − 0.151 to − 0.034; p = 0.0022). In addition, the proportion of time spent hospitalised for respiratory reasons was significantly lower in the nusinersen arm when compared with the SoC arm (least-square mean treatment difference − 8.638%; 95% CI − 14.190 to − 3.086; p = 0.0026) [61]. To adjust costs, the model used the ratio of ventilation use observed in ENDEAR (0.66; 95% CI 0.32–1.37) [22]) as an adjustment factor applied to the proportion of each respiratory care cost item. The ENDEAR rate of hospitalisation (0.759; 95% CI 0.548–1.051 [61]) was used as an adjustment factor applied to the hospitalisation cost item. The proportions between each type of care used in the model (respiratory, gastrointestinal, nutritional or orthopaedic/neurologic) were delimited based on estimated Swedish resource use (Tables S5 and S6 of the ESM).
2.9 Utility Weights
The ENDEAR and CHERISH trials did not collect utility values from patients or caregivers. The economic systematic literature review did not identify utility weights for patients with SMA. Therefore, a vignette study was commissioned to elicit utilities for the health states in the infantile- and later-onset models. In the study, five UK clinical experts reviewed case study descriptions for each health state and were asked to provide an assessment of how health-related quality of life would be affected in each state using the EQ-5D (youth version) [62]. The EQ-5D (youth version) was scored using the EQ-5D-3L UK preference weights. Although it is common in vignette studies that members from the general public rate the severity of the health states, Lloyd et al. [62] used clinical experts because this allowed them to include clinical and health-related quality-of-life information in the descriptions of the health state, and to produce a range of experience of each clinical expert when rating each state. The descriptions of health states were tailored to match the health states in the economic models. The base-case utility weights for the infantile- and later-onset models are presented in Tables 1 and 2, respectively. As the CHERISH study collected the Pediatric Quality-of-Life Inventory (PedsQL™) and a mapping algorithm for PedsQL™ to EQ-5D was available [63], EQ-5D utility values were estimated for the later-onset model health states with the limitation that the patients in the CHERISH trial were substantially younger than the patients used to develop the algorithm [63]. However, these utility values were not explored in the scenario analysis, as they lacked face validity (the utility estimated for the worst health state was 0.73, while the valuation elicited from clinicians in the study by Lloyd et al. [62] for the same health state was −0.13). The model also included disutility for caregivers, as the burden on caregivers of children with SMA is substantial. The impact of using utility data from diseases with similar clinical characteristics was explored in scenario analyses (Tables S7 and S8 of the ESM).
2.10 Adverse Events
No treatment-related adverse events occurred in the ENDEAR trial. In the CHERISH trial, one adverse event experienced by one patient (post-sedation nausea) was considered to be related to treatment. Treatment-related adverse events were not included in the base-case analysis for the two models.
2.11 Base-Case Parameters
Base-case parameters for the infantile- and later-onset models are presented in Tables 1 and 2, respectively.
2.12 Sensitivity and Scenario Analyses
Univariate sensitivity analyses were performed for all parameters to identify those with most influence on the incremental cost-effectiveness ratio (ICER). The probabilistic sensitivity analysis included all parameters (the model was run for 1000 simulations). Estimates of uncertainty were based on the uncertainty in the source data (data permitting). Parameters were sampled from appropriate statistical distributions [64].
2.13 Model Validation
Model validation was performed in alignment with best practices [65] and included face validity (review of model structure and input parameters by two clinical experts and two independent academic health economic experts), internal validity (verification of input data against original sources and programming validation), dependent external validity (comparing model predictions with outcomes in studies used to build the model [7, 47, 50]) and independent external validity (comparing model predictions with outcomes in studies not used to build the model [5, 51, 54, 55]).
3 Results
3.1 Base-Case Deterministic Analyses
The base-case deterministic results for the infantile- and later-onset models are presented in Table 3. In the base-case analysis from the societal perspective, treatment with nusinersen was associated with an ICER (including caregiver disutilities) of approximately 5,635,978 SEK (€551,300) and 3,187,222 SEK (€311,800) per QALY gained for infantile-onset and later-onset SMA, respectively, in comparison to SoC. Treatment with nusinersen was also associated with 3.86 and 9.54 patient incremental QALYs, 0.02 and 2.39 caregiver incremental QALYs, and 6.22 and 1.84 incremental life-years for infantile-onset and later-onset SMA, respectively, when compared to SoC.
3.2 Univariate Sensitivity Analyses
The univariate sensitivity analyses results for the infantile- and later-onset models are presented in Table 4 and Fig. S2 of the ESM. Parameters were varied around the mean value by ±20%. In the infantile-onset model, the health-state utility assigned to the stands/walks unaided health state was the most sensitive parameter, followed by the vial price of nusinersen, producing variations from the base-case ICER (including caregiver disutilities) of − 16% (when the utility was higher) to 23.5% (when the utility was lower) and of − 17.3% to 17.3%, respectively. In the later-onset model, the variations from the base-case ICER of the two most sensitive parameters ranged from − 25.1% to 25.1% and from −10.6% to 13.5% for the vial price and the utility of the walks unaided health state, respectively.
3.3 Probabilistic Sensitivity Analyses
The cost-effectiveness planes and the cost-effectiveness acceptability curve for the infantile- and later-onset models are presented in Fig. 4. The willingness-to-pay threshold at which the probability of cost effectiveness started to be greater than 0% is around 4.2 million SEK/QALY (€410,800) gained for the infantile-onset model and 2.2 million SEK/QALY (€215,200) gained for the later-onset model.

Cost-effectiveness planes for infantile-onset (a) and later-onset (b) spinal muscular atrophy (SMA) and cost-effectiveness acceptability curves for infantile- and later-onset SMA. CEAC cost-effectiveness acceptability curve, PSA probabilistic sensitivity analysis, QALY quality-adjusted life-year, SEK Swedish krona. Note: Results included QALYs accrued by patients and caregivers
3.4 Scenario Analyses
The scenario analyses results are presented in Tables S6 and S7 of the ESM. In the infantile-onset model, the ICER (including caregiver disutilities) ranged from 3,087,460 to 10,194,714 SEK (from €302,000 to €997,200), with the larger variations associated with OS and disease progression assumption after the trial follow-up. The lowest ICERs were observed when alternative external long-term data were used to guide OS (data presented by Zerres and Rudnik-Schoneborn [7] or data from the two other groups of patients receiving respiratory support in the study by Gregoretti et al. [50]). The highest ICER was the result of increasing the risk of death for patients achieving later-onset milestones (i.e. when the adjustment factor was reduced from 0.9 to 0.5). In the later-onset model, the ICERs (including caregiver disutilities) ranged from 1,930,016 to 4,435,635 SEK (from €188,800 to €433,900). The largest ICER was associated with a shorter time horizon of 20 years, followed by increases from the baseline ICERs of 27% and 14.2% when utilities from a similar disease were used (Duchenne muscular dystrophy) and with assumptions regarding disease progression (i.e. 50% of patients receiving nusinersen stop improving and 7% of those patients start progressing as in the SoC arm), respectively. The lowest ICERs were associated with variations to the maintenance dose (i.e. every 6 months as administered in the CHERISH trial).
4 Discussion
The current economic analysis presents the first cost-effectiveness models in the treatment of SMA. The model structure has been used in more than ten health technology assessment submissions across the world, including Sweden [52], England [66], Canada [67], Ireland [68], Scotland [69] and the Netherlands [70]. The models evaluated the cost utility of nusinersen and SoC compared to SoC alone in patients with infantile- and later-onset SMA. Treatment with nusinersen plus SoC was associated with incremental QALYs and incremental costs, translating into ICERs of 5.64 million SEK (€551,300) and 3.19 million SEK (€311,800) per QALY gained in the infantile-onset model and later-onset model, respectively, when compared to SoC. These ICER values are not unusual among similar rare diseases. Schuller et al. [71] present ranges from 2,725,000 SEK to 33,555,000 SEK (from €266,600 to €3,282,300), from 445,000 SEK to 4,422,000 SEK (from €43,500 to €432,600) and from 1,568,000SEK to 10,672,000 SEK (from €153,400 to €1,043,900) for treatments of Fabry, Gaucher and Pompe diseases, respectively.Footnote 2 Landfelt et al. [72] estimated ICERs for Duchenne muscular dystrophy treatments from 16,465,000 SEK to 40,797,000 SEK (€1,610,600 to €3,990,700) for three different modelling approaches.Footnote 3 The TLV does not have a definite willingness-to-pay threshold value; instead it has a range of threshold values that have been reviewed by Svensson et al. [73], which are higher for more severe diseases (could be 40% higher), with the highest cost per QALY of approved reimbursements before nusinersen at 1.22 million SEK (€119,300). The TLV discussed in their reimbursement decision for Cerezyme® (imiglucerase) and Vpriv® (velaglucerase alfa) for the treatment of Gaucher’s disease the set of criteria under which a threshold of 2 million SEK per QALY could be considered reasonable for rare disease [74].
Univariate sensitivity analyses showed that the two parameters most sensitive to change for both models were nusinersen vial price and patient health-state utilities, specifically that of the walking without assistance health states. Probabilistic sensitivity analyses resulted in a mean ICER of 5.68 million SEK (€555,900) [95% credible interval 4.61–7.89 million SEK] in the infantile-onset model and a mean ICER of 3.1 million SEK (€303,600) [95% credible interval 2.4–4.4 million SEK] in the later-onset model. Scenario analysis showed variations from the base-case ICER (including caregivers) from 45 to 81% and from − 39 to 39% in the infantile-onset and later-onset models, respectively.
Study strengths included the conduct of economic and clinical systematic literature reviews to identify model input data and two studies to elicit utility data not available in the literature. Clinical and health economic experts provided input and validated the model structures through interviews and an advisory board. Survival data from the trial were analysed in accordance with health technology assessment guidelines (TLV and National Institute for Health and Care Excellence), and extensive sensitivity analyses were performed. The model was validated in alignment with best practice [65]. However, cross validation was not possible, as this is the first economic evaluation for the treatment of SMA. The model was programmed with sufficient flexibility to be adapted to other country settings.
These economic evaluations, like others in rare diseases, are associated with methodological limitations. The ENDEAR and CHERISH clinical trials had statistically small sample sizes (both trials had 2:1 allocation ratios; ENDEAR, n = 121; CHERISH, n = 126). Furthermore, the CHOP INTEND and HFMSE clinical outcome measures only capture motor functions; therefore, additional treatment benefits, including respiratory improvements, are not fully captured by the economic evaluations. The maintenance dose in the CHERISH trial was given every 6 months; however, the maintenance dose used in the model was every 4 months, as specified by the European Medicines Agency. No efficacy adjustment was performed to account for a more frequent maintenance dose. The ICER was reduced by 39% when the maintenance dose was given every 6 months because of a decrease in the drug costs.
Moreover, there is uncertainty associated with the resource use data, long-term survival and utility weights. Clinician interviews, undertaken to elicit resource use data, were limited just to two clinicians. A healthcare registry-based collection would possibly have provided more accurate resource use estimates for patients with SMA in primary care, outpatient specialist care and inpatient care. However, healthcare registries in Sweden would only provide limited information regarding the resource use for patients with SMA and would not capture items such as assistive devices, personal assistance, transportation, and caregiver production loss. A future cost-effectiveness analysis for the treatment of SMA in Sweden should be supported by resource use data collected from healthcare registries in Sweden complemented with resource use data elicited from clinical experts for data not available in the registries.
A scenario analysis was explored using the resource use and costs from a cross-sectional survey study in Germany, [57] which resulted in an increase of the ICER by 9.5% in the infantile-onset model and by 14.4% in the later-onset model. With 82% of patients in the treatment arm and 58% of patients in the SoC arm still alive at the end of the ENDEAR follow-up and with no deaths reported in the CHERISH trial, the models’ survival predictions were highly sensitive to different treatment effect assumptions and to the choice of external data to guide OS after the trial follow-up. The scenario where 7% of patients (of those who reached an improvement plateau) in the nusinersen arm progressed as patients in the SoC arm resulted in an ICER (including caregivers) increase of 29% in the infantile-onset model and an increase of 14% in the later-onset model. Furthermore, the limitations of the vignette study undertaken by Lloyd et al. [62] increased the uncertainty of the utility weights used in the base-case analysis.
An alternative scenario could be explored in a future cost-effectiveness analysis where the Swedish EQ-5D tariff is used instead of the UK tariff to elicit the utilities from the vignette study. However, the impact of using alternative utility data from similar diseases was evaluated in scenario analyses, which resulted in changes from the base-case ICERs (including caregivers) ranging from − 9.8 to 8.2% in the infantile-onset model and from 11.3 to 27.0% in the later-onset model. Resulting utilities from the PedsQL™ mapping study varied from 0.73 to 0.88 (worst-to-best health state), which lacked face validity and varied widely from utility ranges observed in similar rare diseases [75] and therefore may underestimate the benefit of nusinersen.
In addition, there are some limitations associated with the algorithm used to map the PedsQL™ data, as it was developed for an older population than the population in the CHERISH trial and did not include the neuromuscular module. Further evaluation of the cost effectiveness of nusinersen is recommended when longer term data are available from the ongoing open-label SHINE extension study (NCT02594124), into which patients from ENDEAR and CHERISH were moved, to incorporate additional evidence on treatment benefit into the economic model.
5 Conclusions
The infantile-onset model resulted in 3.86 incremental QALYs for patients, 0.02 incremental QALYs for caregivers and 6.22 incremental life-years with nusinersen treatment plus SoC vs. SoC. The incremental costs were 21,854,051 SEK (€2,137,700), resulting in an ICER (including caregivers) of 5,635,978 SEK (€551,300) per QALY gained. The later-onset model resulted in 9.54 incremental QALYs for patients, 2.39 incremental QALYs for caregivers and 1.84 life-years gained with nusinersen treatment plus SoC vs. SoC. The incremental costs were 38,023,409 SEK (€3,719,400), resulting in an ICER (including caregivers) of 3,187,222 SEK (€311,800) per QALY gained. In scenario analyses, the ICER (including caregivers) ranged from 3,087,460 to 10,194,714 SEK (from €302,000 to €997,200) in the infantile-onset model and from 1,930,016 to 4,435,635 SEK (from €188,800 to €433,900) in the later-onset model. Nusinersen was not cost effective in the infantile- and later-onset populations when using a willingness-to-pay threshold [2 million SEK (€195,600)] that the TLV has considered reasonable for some rare diseases yet nusinersen has gained reimbursement in Sweden in 2017 for patients with SMA types I–IIIa. The models can be adapted to evaluate the cost effectiveness of nusinersen in other countries.
Notes
Based on the Euro-to-SEK exchange rate as of 8/3/2018 and using Google finance converter (1 Euro = 10.223 SEK); rounded to the nearest hundred. The same exchange rate used for values in Euros is given in brackets.
Based on Euro-to-SEK exchange rate as of 8/3/2018 using Google finance converter (1 Euro = 10.223 SEK); rounded to the nearest thousand.
Based on GBP-to-SEK and Euro-to-SEK exchange rate as of 8/3/2018 using Google finance converter (1 GBP = 11.4125 SEK; 1 Euro = 10.223 SEK); rounded to the nearest thousand.
References
Darras BT. Spinal muscular atrophies. Pediatr Clin N Am. 2015;62:743–66.
Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027–49.
Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51:157–67.
Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11:443–52.
Farrar MA, Vucic S, Johnston HM, du Sart D, Kiernan MC. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr. 2013;162:155–9.
Finkel R, Bertini E, Muntoni F, Mercuri E. 209th ENMC international workshop: outcome measures and clinical trial readiness in spinal muscular atrophy, 7–9 November 2014, Heemskerk, the Netherlands. Neuromuscul Disord. 2015;25:593–602.
Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy: clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52:518–23.
Rudnik-Schoneborn S, Hausmanowa-Petrusewicz I, Borkowska J, Zerres K. The predictive value of achieved motor milestones assessed in 441 patients with infantile spinal muscular atrophy types II and III. Eur Neurol. 2001;45(3):174–81.
Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy: recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11:351–9.
Arkblad E, Tulinius M, Kroksmark AK, Henricsson M, Darin N. A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatr. 2009;98(5):865–72.
Jedrzejowska M, Milewski M, Zimowski J, Zagozdzon P, Kostera-Pruszczyk A, Borkowska J, et al. Incidence of spinal muscular atrophy in Poland: more frequent than predicted? Neuroepidemiology. 2010;34(3):152–7.
Prior TW. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med. 2010;12(3):145–52.
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of > 72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32.
Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet. 2004;12(12):1015–23.
Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175–86.
Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, et al. Diagnosis and management of spinal muscular atrophy. Part 1. Recommendations for diagnosis, rehabilitation, orthopaedic and nutritional care. Neuromuscul Disord. 2018;28:103–15.
Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, et al. Diagnosis and management of spinal muscular atrophy. Part 2. Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28:197–207.
Cuisset JM, Estournet B. Recommendations for the diagnosis and management of typical childhood spinal muscular atrophy. Rev Neurol (Paris). 2012;168:902–9.
De Sanctis R, Coratti G, Pasternak A, Montes J, Pane M, Mazzone ES, et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord. 2016;26:754–9.
Davis RH, Godshall BJ, Seffrood E, Marcus M, LaSalle BA, Wong B, et al. Nutritional practices at a glance: spinal muscular atrophy type I nutrition survey findings. J Child Neurol. 2014;29(11):1467–72.
Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet. 2013;6:113–20.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–32.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2017;388:3017–26.
ClinicalTrials.gov. A study to assess the efficacy, safety and pharmacokinetics of nusinersen (ISIS 396443) in infants with spinal muscular atrophy (SMA). 2017. https://www.clinicaltrials.gov/ct2/show/NCT01839656?term=A+Study+to+Assess+the+Efficacy%2C+Safety+and+Pharmacokinetics+of+Nusinersen+%28ISIS+396443%29+in+Infants+With+Spinal+Muscular+Atrophy+%28SMA%29.&rank=1. Accessed 17 Nov 2017.
ClinicalTrials.gov. A study to assess the safety and tolerability of nusinersen (ISIS 396443) in participants with spinal muscular atrophy (SMA) (EMBRACE). 2017. https://clinicaltrials.gov/ct2/show/NCT02462759. Accessed 17 Nov 2017.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378:625–35.
ClinicalTrials.gov. An open-label safety, tolerability and dose-range finding study of multiple doses of nusinersen (ISIS 396443) in participants with spinal muscular atrophy. 2017. https://www.clinicaltrials.gov/ct2/show/NCT01703988?term=NCT01703988&rank=1. Accessed 17 Nov 2017.
ClinicalTrials.gov. An open-label safety and tolerability study of nusinersen (ISIS 396443) in participants with spinal muscular atrophy (SMA) who previously participated in ISIS 396443-CS2 (NCT01703988) or ISIS 396443-CS10 (NCT01780246). 2017. https://www.clinicaltrials.gov/ct2/show/NCT02052791?term=NCT02052791&rank=1. Accessed 17 Nov 2017.
ClinicalTrials.gov. An open-label safety, tolerability, and dose-range finding study of nusinersen (ISIS 396443) in participants with spinal muscular atrophy (SMA) (SMNRx). 2017. https://www.clinicaltrials.gov/ct2/show/NCT01494701?term=NCT01494701&rank=1. Accessed 17 Nov 2017.
ClinicalTrials.gov. An open-label safety and tolerability study of nusinersen (ISIS 396443) in participants with spinal muscular atrophy who previously participated in ISIS 396443-CS1 (NCT01494701). 2017. https://www.clinicaltrials.gov/ct2/show/NCT01780246?term=NCT01780246&rank=1. Accessed 17 Nov 2017.
ClinicalTrials.gov. A study of multiple doses of nusinersen (ISIS 396443) delivered to infants with genetically diagnosed and presymptomatic spinal muscular atrophy (NURTURE). 2017. https://clinicaltrials.gov/ct2/show/NCT02386553. Accessed 17 Nov 2017.
ClinicalTrials.gov. A study for participants with spinal muscular atrophy (SMA) who previously participated in nusinersen (ISIS 396443) investigational studies. (SHINE). 2017. https://clinicaltrials.gov/ct2/show/NCT02594124. Accessed 17 Nov 2017.
Haataja L, Mercuri E, Regev R, Cowan F, Rutherford M, Dubowitz V, et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr. 1999;135:153–61.
Glanzman AM, Mazzone E, Main M, Pelliccioni M, Wood J, Swoboda KJ, et al. The children’s hospital of philadelphia infant test of neuromuscular disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20:155–61.
O’Hagen JM, Glanzman AM, McDermott MP, et al. An expanded version of the Hammersmith functional motor scale for SMA II and III patients. Neuromuscul Disord. 2007;17:693–7.
Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, et al. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One. 2010;5:e12140.
WHO Multicentre Growth Reference Study Group. WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl. 2006;450:86–95.
Mazzone ES, Mayhew A, Montes J, Ramsey D, Fanelli L, Young SD, et al. Revised upper limb module for spinal muscular atrophy: development of a new module. Muscle Nerve. 2017;55:869–74.
US Food and Drug Administration. FDA news release: FDA approves first drug for spinal muscular atrophy. 2016. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm534611.htm. Accessed 17 Nov 2017.
European Medicines Agency. Spinraza (nusinersen). 2017. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004312/human_med_002119.jsp&mid=WC0b01ac058001d124. Accessed 17 Nov 2017.
Pearson I, Rothwell B, Olaye A, Knight C. Economic modeling considerations for rare diseases. Value Health. 2018;21(5):515–24.
Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83:810–7.
Mercuri E, Finkel R, Montes J, Mazzone E, Sormani MP, Main M, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord. 2016;26:126–31.
McGraw S, Qian Y, Henne J, Jarecki J, Hobby K, Yeh WS. A qualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. 2017;17(1):68.
Pera MC, Coratti G, Forcina N, Mazzone ES, Scoto M, Montes J, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017;17(1):39.
Pharmaceutical Benefits Board. General guidelines for economic evaluations from the Pharmaceutical Benefits Board (LFNAR 2003:2). 2003. https://www.tlv.se/in-english/medicines/apply-for-reimbursement.html. Accessed 21 Nov 2017.
Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146:67–72.
Spinal Muscular Atrophy Support UK. SMA drug pipeline. 2018. http://www.smasupportuk.org.uk/drug-pipeline. Accessed 15 Aug 2018.
Latimer N. NICE DSU technical support document 14. Survival analysis for economic evaluations alongside clinical trials: extrapolation with patient level data. 2013. http://www.nicedsu.org.uk/NICE%20DSU%20TSD%20Survival%20analysis.updated%20March%202013.v2.pdf. Accessed 5 May 2017.
Gregoretti C, Ottonello G, Chiarini Testa MB, Mastella C, Rava L, Bignamini E, et al. Survival of patients with spinal muscular atrophy type 1. Pediatrics. 2013;131:e1509–14.
Oskoui M, Levy G, Garland CJ, Gray JM, O’Hagen J, De Vivo DC, et al. The changing natural history of spinal muscular atrophy type 1. Neurology. 2007;69:1931–6.
Tandvårds-och läkemedelsförmånsverkets författningssamling. Spinraza (nusinersen) intended for the treatment of spinal muscle atrophy of type 5q. 2017. https://www.tlv.se/download/18.73f5568815f9b31688a88ff4/1510556738560/halsoekonomisk_bedomning171108_underlag_spinraza.pdf. Accessed 21 Feb 2018.
Lawson CHR. Solving least squares problems. Englewood Cliffs: Prentice-Hall; 1974.
Ge X, Bai J, Lu Y, Qu Y, Song F. The natural history of infant spinal muscular atrophy in China: a study of 237 patients. J Child Neurol. 2012;27:471–7.
Mannaa MM, Kalra M, Wong B, Cohen AP, Amin RS. Survival probabilities of patients with childhood spinal muscle atrophy. J Clin Neuromuscul Dis. 2009;10:85–9.
Mogensen UB, Ishwaran H, Gerds TA. evaluating random forests for survival analysis using prediction error curves. J Stat Software. 2012;50:1–23.
Klug C, Schreiber-Katz O, Thiele S, Schorling E, Zowe J, Reilich P, et al. Disease burden of spinal muscular atrophy in Germany. Orphanet J Rare Dis. 2016;11(1):58.
López-Bastida J, Pena-Longobardo LM, Aranda-Reneo I, Tizzano E, Sefton M, Oliva-Moreno J. Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis. 2017;12:141.
Apoteket. https://www.apoteket.se/produkt/spinraza-injektionsvatska-losning-12-mg-1-x-5-milliliter-injektionsflaska-534913/. Accessed 2 Apr 2017.
Södra Regionvårdsnämnden. Regional prices and refund rates for the Southern health care region, 2018. 2018. http://sodrasjukvardsregionen.se/avtal-priser/regionala-priser-och-ersattningar/. Accessed 21 Nov 2018.
Tulinius M, Bruno C, Deconinck N, Schara U, Finkel RS, Bhan I, et al. Nusinersen decreases the incidence and length of hospitalisations in infants with spinal muscular atrophy: results from the ENDEAR study. Poster presented at SMA Europe’s International Scientific Congress on SMA; Kraków; 25–27 Jan 2018.
Lloyd AJ, Gallop K, Thompson R, Vaidya S, Teynor M. Estimation of the health-related quality of life benefits of treatment for spinal muscular atrophy (SMA). Poster presented at the ISPOR 20th Annual European Congress; Glasgow; 4–6 Nov 2017.
Khan KA, Petrou S, Rivero-Arias O, Walters J, Boyle SE. Mapping EQ-5D utility scores from the PedsQL™ generic core scales. Pharmacoeconomics. 2014;32:693–706.
Briggs A. Probabilistic analysis of cost-effectiveness models: statistical representation of parameter uncertainty. Value Health. 2005;8:1–2.
Eddy DM, Hollingworth W, Caro JJ, Tsevat J, McDonald KM, Wong JB. Model transparency and validation: a report of the ISPOR-SMDM modeling good research practices task force-7. Value Health. 2012;15:843–50.
National Institute for Health and Care Excellence. Single technology appraisal: nusinersen for treating spinal muscular atrophy [ID1069]. Committee papers. Manchester: National Institute for Health and Care Excellence; 2018. https://www.nice.org.uk/guidance/gid-ta10281/documents/committee-papers.
Canadian Agency for Drugs and Technologies in Health. Pharmacoeconomic review report: nusinersen for treatment of patients with 5q SMA. Canadian Agency for Drugs and Technologies in Health; 2018. https://www.ncbi.nlm.nih.gov/books/NBK534118/pdf/Bookshelf_NBK534118.pdf.
National Centre for Pharmacoeconomics. Cost-effectiveness of nusinersen (Spinraza) for the treatment of 5q spinal muscular atrophy (SMA). National Centre for Pharmacoeconomics; 2017. http://www.ncpe.ie/wp-content/uploads/2017/08/Summary-Nusinersen.pdf.
Scottish Medicines Consortium. Nusinersen for the treatment of 5q spinal muscular atrophy (SMA). Glasgow: Scottish Medicines Consortium (SMC); 2018. https://www.scottishmedicines.org.uk/media/3415/nusinersen-spinraza-final-april-2018-for-website.pdf.
Zorginstituut Nederland. Package advice nusinersen (Spinraza®) for the treatment of spinal muscular atrophy (SMA). 2018. https://www.zorginstituutnederland.nl/publicaties/adviezen/2018/02/07/pakketadvies-nusinersen-spinraza-voor-de-behandeling-van-spinale-musculaire-atrofie-sma. Accessed 15 Jun 2018.
Schuller Y, Hollak CE, Biegstraaten M. The quality of economic evaluations of ultra-orphan drugs in Europe: a systematic review. Orphanet J Rare Dis. 2015;10:92.
Landfeldt E, Alfredsson L, Straub V, Lochmüller H, Bushby K, Lindgren P. Economic evaluation in Duchenne muscular dystrophy: model frameworks for cost-effectiveness analysis. Pharmacoeconomics. 2017;35:249–58.
Svensson M, Nilsson FO, Arnberg K. Reimbursement decisions for pharmaceuticals in Sweden: the impact of disease severity and cost effectiveness. Pharmacoeconomics. 2015;33(11):1229–36.
Tandvårds-och läkemedelsförmånsverkets. Underlag för beslut om subvention: Cerezyme och Vpriv. https://www.tlv.se/download/18.467926b615d084471ac33d95/1510316366212/Bes161219_underlag_for_beslut_cerezyme_vpriv.pdf. Accessed 21 Feb 2018.
Teynor M, Thompson R, Lundqvist T, Ekelund M, Zuluaga-Sancez S, Knight C. Benchmarking utility estimates in rare diseases may guide the selection of data inputs in health economic modeling. Poster presented at the 9th European Conference on Rare Diseases and Orphan Products; Vienna; 10–12 May 2018.
Burström K, Johannesson M, Diderichsen F. Swedish population health-related quality of life results using the EQ-5D. Qual Life Res. 2001;10:621–35.
Acknowledgements
The authors acknowledge the team that performed the systematic literature review: Emily Moss, Rikal Bhaila and Louise Hartley. The authors also acknowledge Gail Zona for editorial services and Tiina Carlsson, formerly employed as Scientist at Nordic Health Economics AB, for compiling the resource use and cost report regarding SMA.
Author information
Authors and Affiliations
Contributions
Santiago Zuluaga Sanchez and Christopher Knight designed the cost-effectiveness models with input from Megan Teynor, Robin Thompson, Thomas Lundqvist and Mats Ekelund. Adrian D. Vickers conducted the survival analysis used in the models. Annabelle Forsmark led the collection of resource use data used to estimate the costs in the models. Andrew Lloyd led the study eliciting the health utility values used in the models. Santiago Zuluaga Sanchez developed the cost-effectiveness models and executed all analyses. Santiago Zuluaga Sanchez led the interpretation of findings together with Christopher Knight, Thomas Lundqvist, Mats Ekelund and Annabelle Forsmark with input from the other authors. Santiago Zuluaga Sanchez and Annabelle Forsmark drafted the manuscript. All authors reviewed the manuscript and approved the decision to submit for publication.
Corresponding author
Ethics declarations
Funding
Open access fees were funded by Biogen Inc. This study was funded by Biogen Inc.
Conflict of Interest
Santiago Zuluaga Sanchez, Christopher Knight and Adrian D. Vickers are full-time employees of RTI Health Solutions, which received funding from Biogen Inc. to conduct this study. The contract between RTI Health Solutions and the sponsor includes independent publication rights. RTI Health Solutions conducts work for the government, public and private organisations, including pharmaceutical companies. Robin Thompson, Thomas Lundqvist and Mats Ekelund are employees of Biogen Inc. Megan Teynor is a former employee of Biogen Inc. Annabelle Forsmark is a full-time employee of Nordic Health Economics AB, which received funding from Biogen Inc. for contributing to this study. Nordic Health Economics AB conducts work involving health economics and outcomes research for the government, public and private sectors. Andrew Lloyd is a Director of Acaster Lloyd Consulting Ltd, which received funding from Biogen Inc. for work that supported the modelling exercise.
Data Availability
The model generated during the current study is not publicly available because it includes patient-level data from the ENDEAR and CHERISH clinical trials that could be deanonymized.
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to Participate
Informed consent procedures were not applicable to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zuluaga-Sanchez, S., Teynor, M., Knight, C. et al. Cost Effectiveness of Nusinersen in the Treatment of Patients with Infantile-Onset and Later-Onset Spinal Muscular Atrophy in Sweden. PharmacoEconomics 37, 845–865 (2019). https://doi.org/10.1007/s40273-019-00769-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-019-00769-6