
Abstract
Introduction
To evaluate the possibility that switching from reference biologic medicines to biosimilars could lead to altered clinical outcomes, including enhanced immunogenicity, compromised safety, or diminished efficacy for patients, a systematic literature review was conducted of all switching studies between related biologics (including biosimilars).
Methods
A systematic search was conducted using the Medline® and Embase® databases up to 30 June 2017 employing specific medical subject heading terms. Additionally, the snowball method and a hand search were also applied. Publications were considered if they contained efficacy or safety information related to a switch from a reference medicine to a biosimilar. Non-English, non-human studies, editorials, notes, and short surveys were excluded.
Results
Primary data were available from 90 studies that enrolled 14,225 unique individuals. They included protein medicines used in supportive care as well as those used as therapeutic agents. The medicines contained seven different molecular entities that were used to treat 14 diseases. The great majority of the publications did not report differences in immunogenicity, safety, or efficacy. The nature and intensity of safety signals reported after switching from reference medicines to biosimilars were the same as those already known from continued use of the reference medicines alone. Three large multiple switch studies with different biosimilars did not show differences in efficacy or safety after multiple switches between reference medicine and biosimilar. Two publications reported a loss of efficacy or increased dropout rates.
Conclusions
While use of each biologic must be assessed individually, these results provide reassurance to healthcare professionals and the public that the risk of immunogenicity-related safety concerns or diminished efficacy is unchanged after switching from a reference biologic to a biosimilar medicine.
Similar content being viewed by others

Scientific literature (1993 up to 30 June 2017) was reviewed to identify publications that contained primary data on single or multiple switching from reference biological medicines to biosimilars. |
A total of 90 studies were identified involving seven molecular entities that treated 14 disease indications, and enrolled a total of 14,225 individuals. |
The great majority of studies did not report differences in safety, efficacy, or immunogenicity after a single switch event compared to patients that were not switched. Only a small number (three) of multiple switch studies have been published to date, but likewise no differences were detected. |
Overall, the results suggest a low risk of either a safety concern or a loss of efficacy after switching to a biosimilar. |
1 Introduction
Biological medicines (biologics) are medicines made in living systems. Biosimilars are copies of already licensed biologics (referred to as the reference medicine) that are highly similar, but that are made by different sponsors using independently-derived cell lines and separately-developed manufacturing processes [1, 2]. Biosimilars can only be approved if a manufacturer demonstrates that there are no clinically meaningful differences in safety, efficacy, and immunogenicity when directly compared with the reference medicine [3].
The experience with the reference medicine, in both pre-approval clinical trials and post-approval routine clinical practice of medicine, provides a baseline for the safety and efficacy expected for both reference medicines and their corresponding biosimilars. To date, no new safety or efficacy concerns have been detected in the over 10 years and greater than 700 million days of patient experience with biosimilars [4].
Nonetheless, concerns have been raised that switching patients from reference medicines to biosimilars, or other structurally-related biologics, may lead to increased immunogenicity and consequential safety problems, or even a loss of efficacy. A review of switching studies reported in the literature is an important first step to confirm or deny any existing pattern that may exist related to biologic switching. Switches occur when patients receive medicines formally designated as biosimilars, but may also occur after manufacturing process changes have occurred, if the process changes lead to structural modifications or changes in the impurity profile of the biologic drug [5].
A commonly expressed concern is whether there is an increase in immunogenicity related to the act of switching itself. Anti-drug antibody (ADA) assays likely offer the most sensitive method to detect immunogenicity; and neutralizing antibodies (NAB) assays are the most direct method to signal the potential clinical relevance of ADAs. Pharmacokinetics, efficacy and certain safety events may be additional measures to detect clinically relevant immunogenicity.
Several product class or treatment specific literature reviews of switching studies from reference medicines to biosimilars have been published [6,7,8]. The aim of this systematic literature review was to provide a single survey that includes all switching studies of biologics to biosimilars, providing a baseline in one manuscript of all switching studies published prior to 30 June 2017.
2 Methods
2.1 Data Sources and Searches
A systematic search using the Medline® and Embase® databases up to 30 June 2017 was conducted. Medical subject heading (MeSH) terms like “biosimilar pharmaceuticals” OR “biologic factors” were employed. A biological drug was included if a copy version of the reference medicine was approved in either the USA or EU as a biosimilar. Additional MeSH terms of all the smaller and larger protein biologics (erythropoietin, human growth hormone, filgrastim, etanercept, adalimumab, infliximab, and rituximab) were added to the search string. This string was combined with the MeSH term “drug substitution.” As the medical subject heading “biosimilar” was first introduced by National Center for Biotechnology Information in 2012 [9], further references within eligible papers were also scrutinized for related evidence with the “snowball method” [10, 11]. Additional hand search was applied through citation review of identified articles.
2.2 Study Selection
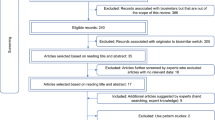
Publications were considered if they contained efficacy or safety information related to a switch from a reference medicine to a biosimilar. Inclusion criteria were as broad as possible to maximize capture of available data on switching. Randomized clinical trials (RCTs) and observational studies that provide real world evidence (RWE) were both included because they provide useful and complementary information. Non-English, non-human studies, editorials, notes, and short surveys were excluded. Switching studies from erythropoietin to darbepoetin; erythropoietin to pegylated-erythropoietin; and insulin to insulin were excluded as they were out of scope of this review. An experienced researcher was involved in an abstract screening and subsequent full text screening steps to arrive at articles with primary data for in depth literature review. An assessment of study methods was not considered as a screening parameter for a given publication. Figure 1 describes the step-by-step citation disposition.

Citation disposition for the literature search
2.3 Data Extraction and Quality Assessment
The selected full text articles were extracted for various parameters such as study design, patient demographics, safety, efficacy, immunogenicity, and adverse events (AEs) and imported into MS Excel by one researcher and verified by another researcher. Multiple reports from the same study were identified by an experienced researcher by employing methodology from the Cochrane Community [12]. Various measures such as common author names, date, and duration of the study, sample size, study location, and setting were compared to detect such reports. As a next step, extracted data from multiple reports of the same study were linked together in an MS-Excel database and the collated information for a particular study is presented in the Supplementary Material tables. Citation of a full-text article with complete study data was preferred over abstracts published at an earlier date. In case of multiple abstracts describing results from a single study but that contained mutually exclusive data, an abstract was cited in tables that contributed the most relevant data. Due to variability in individual studies, a cross-study quality assessment was not feasible and was therefore not conducted.
2.4 Data Synthesis and Analysis
The evidence tables (Tables 2 and 3) were the outcome of the data synthesis and analysis step. The objective of these analyses was to evaluate the possibility that switching from reference biologic medicines to biosimilars could lead to altered clinical outcomes. Tables were organized by smaller and larger biologic proteins and disease indication. Indication-wise safety, efficacy, immunogenicity, and AE data are presented in supplementary tables (Supplemental Tables 1–11). Because of varying study designs, endpoints, and statistical methodologies employed; no effort was made to conduct a meta-analysis. Instead, all endpoints are reported in a descriptive manner in order to be inclusive.
3 Results
A systematic search resulted in 2,045 citations. As shown in Fig. 1, after applying the exclusion criteria, 1,127 citations remained for the abstract screening step. After this initial screening, 365 citations were short-listed for full text screening. Short-listed full texts were scanned to select 151 articles for in depth literature review.
The molecular characteristics of the biologics varied along a continuum as opposed to discrete categories (Table 1) limiting the ability to correlate protein complexity with any conclusions that might be drawn about safety or efficacy. Biologics were arbitrarily divided into smaller proteins and larger proteins for the sake of simplicity. This yielded 57 studies of small protein biologics (e.g., erythropoietin, filgrastim, human growth hormone) as well as 94 studies of larger biologics (proteins ≥ 200 amino acids length, including fusion proteins [etanercept] and monoclonal antibodies [adalimumab, infliximab and rituximab]) that contained primary safety, efficacy, immunogenicity and AE data.
Of the 94 selected articles or abstracts involving the larger proteins, 54 contained primary data. The others were either review articles, multiple reports from the same study (N = 30) or briefing documents from regulatory bodies (N = 10).
In addition to reference medicine to biosimilar switching studies in smaller protein biologics, we identified 14 studies that enrolled 5,256 patients who were switched from erythropoietin to darbepoetin. We also identified five studies that evaluated switching from erythropoietin to pegylated-erythropoietin that enrolled 1,941 patients (data not shown). But since darbepoetin and pegylated-erythropoietin are versions of erythropoietin in which structural modifications were made to extend product half-life, we view these as switches between different products, and as such these studies are out of scope of this review. In this regard, we differ from an earlier review of smaller proteins that included switching from originator biologics to both biosimilars and proteins of the same product class [8]. Two studies each reported unique data in two different publications; we only considered each study once. After exclusion of these studies, there were 36 studies of smaller proteins that contained switching data.
We identified 46 insulin-to-insulin switching studies that enrolled over 20,000 patients (data not shown). These studies were excluded from the review and analysis because all insulin switching studies were conducted with drugs approved as unique biologics and not as biosimilars.
Altogether, there were 90 studies of both smaller and larger proteins that enrolled 14,225 unique individuals and that contained primary switching data (Table 2; full references are provided in Supplemental Table 12). They included seven different molecular entities used to treat 14 diseases. Safety, efficacy, or immunogenicity endpoints were incorporated into all studies, but only a limited number of studies included all three categories. The exact efficacy endpoints depended on the disease being treated. All safety endpoints were descriptive. There has been a steady growth in this literature from 1993 to the present with rapid growth in 2015 and 2016 (Fig. 2). Seven switching studies evaluated the use of a single biosimilar in multiple patient populations who were being treated individually for a variety of clinical conditions (Supplemental Table 1).

Growth in publications related to biosimilar switching, from 1993 to 2016
The majority of the switching studies enrolled 30–60 subjects, although 33 studies (13 smaller and 20 larger biologics) enrolled more than 100 subjects each. Eight studies enrolled fewer than 20 patients but were included for completeness and also because they often contained patient-specific information. There were no published reports switching from one biosimilar to another biosimilar. Both RCTs and RWE are available in the literature with information on switching from reference medicines to their corresponding biosimilar (Fig. 3). RCTs provide detailed information from controlled clinical experiments that apply specific inclusion and exclusion criteria, while RWE studies provide data from all patients that utilize a given drug, without the inclusion and exclusion criteria limitations applied to the RCTs.

Types of study designs across larger and smaller biologics. Biosimilars of adalimumab, etanercept, and rituximab were approved as of the cut-off date for manuscript. However, there was likely insufficient time for completion and publication of RWE with etanercept and rituximab, and a biosimilar adalimumab was not marketed as of the cut-off date. RCT randomized clinical trial, RWE real world evidence, rHuEPO recombinant human erythropoietin, GH growth hormone
Many of the studies were pharmacokinetic (PK) or pharmacodynamic (PD) studies conducted in healthy subjects. As is common for PK and PD studies, these studies enrolled smaller numbers of subjects than efficacy and safety studies. However, confirmatory clinical efficacy and safety studies in patients were often conducted as a part of biosimilar development programs. Thirty-six publications provided primary data describing efficacy of the larger biologics after switching from reference medicines to the corresponding biosimilars (Table 3). Of these, 12 (33%) were single arm studies describing patients that were switched and the other 24 (67%) were cohort studies comparing switched versus non-switched patients. In the vast majority of these studies, overall efficacy was comparable in maintenance versus the switched groups, or was maintained before and after the switching event in the “cohort studies.” Sporadic observations of loss of responses were reported in a few studies, such as Kang et al. [13], who reported loss of efficacy in one of 17 patients in their study, but no consistent pattern occurred.
Multiple reports published of larger biologics focused on safety endpoints, including AEs known to be associated with reference medicines. Of the switching articles we examined, treatment-emergent adverse events (TEAEs) and treatment-emergent serious adverse events (TESAEs) were reported in 39 studies (Table 3). An active switch design was used in N = 13/39 studies (33%), while a cohort design was employed in N = 24/39 of studies (62%). Serious adverse events (SAEs) were reported as “Nil” (0%) in 32% of the studies.
The percentage of TEAEs and TESAEs in switch and reference arms were comparable across disease indications in seven articles that reported values for larger biologics.
There were 24 studies assessing ADAs in larger biologics with seven also providing information on NABs (Table 3, Supplemental Table 2). In the smaller biologics, 13 studies assessed ADAs of which four studies also reported data on NABs. Diverse methods were used to assess ADAs including radio-immunoassays, enzyme-linked immunosorbent assays (ELISA), and electroluminescence assays. ADA rates varied depending on the type of assay used, often for the same molecule. In all studies reporting immunogenicity data, ADA and NAB levels were found to be comparable at baseline and at the end of study across all disease indications and treatment groups.
The Norwegian government sponsored a switching study to assess safety, efficacy, and immunogenicity of patients switched to biosimilar infliximab from the reference medicine. The NOR-SWITCH study was a randomized, non-inferiority, double-blinded, phase IV trial in patients with immune-mediated inflammatory diseases (Crohn’s disease (CD), ulcerative colitis (UC), psoriasis (PsO), psoriatic arthritis (PsA), rheumatoid arthritis (RA) and spondyloarthritis (SpA)) [14]. A total of 482 patients who were on stable treatment with reference infliximab for at least 6 months were randomized to either infliximab reference medicine or a biosimilar (CT-P13). Of the patients enrolled, 155 patients (37%) had CD and 91 (19%) patients had UC. CT-P13 was non-inferior to the reference medicine in respect to efficacy, safety, and immunogenicity for the total study population. The study was not powered to do sub-group analyses for individual diseases.
One report raised safety concerns after switching from reference medicines to a biosimilar. Yazici et al. [15] reviewed a Turkish claims database where 148 patients were switched from reference infliximab to biosimilar infliximab. They reported an 82% drop-out rate in patients who were switched compared to a 24% drop-out rate in control patients that remained on reference infliximab. It is possible that these were chance results because no such large differences in drop-out rates were seen in switched versus control patients in the 46 other studies that evaluated switching between these same biologics.
3.1 Switching Evidence from Arthritis Studies
Switching studies were conducted in multiple arthritic conditions (Table 3, Supplemental Tables 2 and 3). Efficacy endpoints included disease activity scores and ACR20/50/70 (ACR, American College of Rheumatology, 20% response, 50% response, or 70% response to a composite score) [16] to measure percent improvement in tender or swollen joints after predefined time intervals ranging from 14 to 102 weeks. Of note, switching data were recently provided for two separately-developed infliximab biosimilar antibodies approved for arthritic indications: CT-P13 and SB2.
Regulatory approval for CT-P13 was supported with two RCTs in two different indications, each of which incorporated a switching event and at least 1-year follow-up [17, 18]. The first study was a phase I, randomized, double-blinded study comparing PK, safety, and efficacy of biosimilar and reference medicine in patients with AS. The second study was a phase III double-blinded study that evaluated safety and efficacy of CT-P13 and reference medicine in patients with active rheumatoid arthritis co-administered with methotrexate. There were no clinically meaningful differences in safety and efficacy of CT-P13 compared to reference medicine. Comparable immunogenicity was observed in patients with RA or AS who switched from reference medicine to CT-P13.
SB2 approval was supported by a phase III, randomized, double-blinded, parallel group study, comparing it to infliximab reference medicine in 584 patients with moderate-to-severe RA with co-administration of methotrexate [19]. Patients were randomized 1:1 to receive either SB2 or reference medicine, with data available from 290 patients at the end of the primary endpoint readout. After 54 weeks of treatment, patients treated with reference medicine were switched to SB2 and followed to 78 weeks. No changes were detected in safety or efficacy.
An ongoing Danish registry study, DANBIO, is investigating switching from the reference infliximab to CT-P13 in patients with arthritic conditions [20]. Clinical outcomes of patients with RA, PsA, and axial spondylitis who switched from the reference medicine to the biosimilar were investigated. After 12 months, 802 patients were switched to the biosimilar; efficacy was consistent with historical controls on the reference medicine. The observed dropout rate of 16% was comparable to the rate observed with historical controls of the reference medicine.
3.2 Switching Evidence from Inflammatory Bowel Disease (IBD) Studies
Relatively few RCTs were conducted in IBD indications to support registration of biosimilars. Prospective and retrospective cohort studies (Supplemental Table 5) and one RCT that included IBD patients (the NOR-SWITCH study, discussed above) provided switching data that showed no significant difference in efficacy, safety, and immunogenicity when comparing IBD patients that were switched to biosimilars with those that were treated continuously with reference medicines. Efficacy measures included a variety of disease activity indices (e.g., CD activity index, Harvey-Bradshaw index, Lichtiger’s Index Score, pediatric Crohn’s disease activity index, pediatric ulcerative colitis activity index and simple clinical colitis activity index).
Fiorino et al. [21] conducted a prospective, multi-center cohort study of CT-P13 (biosimilar infliximab) that enrolled 313 CD and 234 UC patients. Patients were either naïve to anti-tumor necrosis factor inhibitors (anti-TNF) biologics (group A, N = 311), previously exposed to one or more anti-TNF biologics (group B, N = 139), or previously exposed to the reference version of infliximab (group C, N = 97). Those previously exposed to one or more anti-TNF biologics were on a median drug holiday of 9 months for infliximab and 10 months for other anti-TNF biologics. The remaining 97 patients on infliximab were switched directly to CT-P13. The authors evaluated effectiveness and safety parameters and reported outcomes comparable to previous experience with reference infliximab, although no direct comparisons were performed.
3.3 Switching Evidence from Psoriasis Studies
Five switching studies were conducted in psoriasis (Table 3, Supplemental Table 6). Efficacy was assessed using psoriasis area severity index (PASI) [22] and a visual analogue scale (VAS). Psoriasis is considered a sensitive indication for evaluation of anti-TNF biosimilar medications because skin responses to treatment are relatively rapid with results that can be easily accessed and quantified; there is a large treatment effect size; dosing falls in the linear phase of the dose response, which allows detection of small differences in efficacy should they exist; and biologics used to treat psoriasis are used as monotherapy, avoiding complications associated with concurrent use methotrexate and other disease-modifying anti-rheumatic drugs [23].
Psoriasis phase III clinical confirmation studies were conducted to support approval of biosimilar etanercept (GP2015) and biosimilar adalimumab (ABP-501). For GP2015, the sponsor conducted a phase III clinical confirmation study in patients with moderate-to-severe chronic, plaque-type psoriasis that incorporated three switches [24]. The safety, efficacy, and immunogenicity profiles of the switched and non-switched arms were similar. It is one of the three multiple-switch studies published to date and is discussed in detail below in the section on multiple switching studies.
The phase III clinical confirmation study conducted with ABP-501 in psoriasis patients treated 347 patients, with 174 treated with ABP-501 and 173 with reference medicine [25]. After 16 weeks, the patients in the reference medicine arm of the study were randomized 1:1 to either continue reference medicine or to receive ABP-501. Patients were then followed for 52 weeks. No increase in AEs was observed and the incidence of ADAs remained unchanged following switches from reference medicine to ABP-501.
3.4 Switching Evidence with Smaller Biologics, Including Hematology–Oncology Therapies
Nineteen reports were identified that provided efficacy data after switching from reference medicines to biosimilars in smaller biologics (Table 3). These included filgrastim, human growth hormone, and erythropoietin
Switching between the reference medicine and biosimilar erythropoietins was studied in multiple indications, including chronic kidney disease (CKD), end-stage renal disease (ESRD), and hemodialysis (HD) (Supplemental Tables 7 and 8). The most common indication studied with these drugs was CKD.
Of the seven CKD, two ESRD, and four HD endpoint studies with erythropoietins, the most common efficacy endpoint was a change in hemoglobin levels (Hb) over time. Efficacy results were similar in all erythropoietin studies, including stable mean Hb levels in the ESRD studies.
Two switching studies were conducted using filgrastim (Supplemental Table 9), using changes in absolute neutrophil count (ANC) and incidence of febrile neutropenia (NP) as efficacy endpoints [26, 27]. The largest was a phase 3 clinical confirmation study between a filgrastim biosimilar (EP2006) and its reference [28]. This was a randomized, double-blinded study in which 218 breast cancer patients who received neoadjuvant myelosuppressive chemotherapy were randomized in a double-blinded setting to receive either a biosimilar or the reference medicine. The study incorporated five switches and is discussed in more detail below.
Multiple human growth hormones are available in the EU and US for rectifying deficiencies in human growth hormone levels (GHD). Nine switching studies with human growth hormones were published, with a cumulative total of 470 patients (Table 3). Three of the studies enrolled more than 100 patients each (Table 2). Efficacy endpoints in GHD switching studies included height, height standard deviation scores, and height velocity standard deviation. Comparable efficacy and safety were seen after switching from reference medicines to biosimilars in the seven GHD studies that provided efficacy and safety data (Supplemental Table 10).
3.5 Switching Studies Conducted in Healthy Volunteers
The literature review revealed ten switching studies conducted in healthy volunteers (HV), using filgrastim, erythropoietin, and human growth hormone medicines (Supplemental Table 11). Health authorities recommend conducting pharmacokinetic or immunogenicity studies in healthy adults because they are immunocompetent and have physiological responses that are not compromised by disease conditions or concomitant medications. As a result, comparative studies in HVs may have greater potential to detect differences in clinical response, should any exist, compared to patients being treated for disease indications. In all the switching studies conducted in HVs, safety profiles of individuals who received reference medicines and then biosimilars were similar to safety profiles of individuals who remained on reference medicines.
3.6 Multiple Switch Studies
As of 30 June 2017, only three multiple switch studies have been published, one with a biosimilar filgrastim, the second with a biosimilar etanercept and the third with a biosimilar adalimumab. The third multiple switch study was published after the data cut-off and so is not included in the tally of studies or patients. However, given the paucity of published multiple switching studies and the importance of such studies to questions related to switching and immunogenicity, we elected to include the third multiple switching study in the description of results.
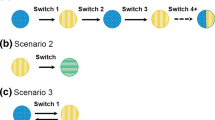
In support of their biosimilar filgrastim registration, the sponsor conducted a clinical safety and efficacy study that incorporated five switchover events (Fig. 4), comparing 107 breast cancer patients that had been switched with 51 patients that had been treated continuously with either the reference medicine or the biosimilar [28]. The results showed no differences in efficacy or overall safety over the course of the study. No NABs were detected in either arms of the study.

Switching study design for biosimilar filgrastim (EP2006) versus reference medicine. d day, DSN duration of severe neutropenia
A cross-over study design incorporating three switchover events (Fig. 5) was incorporated into the clinical safety and efficacy study that supported licensure of a biosimilar etanercept, GP2015 [24]. This was a phase III clinical confirmation study in patients with moderate-to-severe chronic plaque psoriasis. In the initial phase of this study, 264 patients received GP2015 and 267 received the reference medicine. After 12 weeks, each of the two arms was further randomized 2:1 to continue the same treatment or to receive the other medicine. The switched arms were then switched several more times so that after 52 weeks of follow-up, data were available from 178 patients who were switched three times and from 274 patients who remained on the same therapy throughout the study. The safety, efficacy, and immunogenicity profiles of the switched and non-switched arms were similar.

Switching study design for biosimilar etanercept (GP2015) versus reference medicine. wk week
A third multiple switching study was published after the cut-off date for this review. Biosimilar adalimumab (GP2017) and reference medicine were compared in 231 adalimumab biosimilar versus 234 reference medicine patients using a four-switchover study design similar to that used to evaluate biosimilar etanercept (GP2015) in moderate-to-severe chronic plaque psoriasis patients. Efficacy, AEs and immunogenicity were similar for both drugs after 51 weeks [29].
4 Discussion
This literature review was undertaken to see if switching studies with biologics support or do not support the hypothesis that the act of switching from reference medicines to biosimilars is associated with altered immunologic responses. Of note, many biologics have been used for many years and have undergone multiple manufacturing changes, which constitute de facto switches [30, 31]. However, the literature we reviewed did not address these as switches per se. That is, any individual biologic is considered unchanged throughout its lifetime for the purposes of the literature on switching.
Some, but not all, therapeutic proteins are inherently immunogenic. Immunologic responses induced during treatment with therapeutic biologics and their clinical significance may be influenced by a wide variety of factors, including medicine features, patient variables and treatment parameters [32]. For example, infliximab and adalimumab are both known to induce ADAs [33].
The most direct measure of immunogenicity is by use of validated immunoassays. Thirty-seven of the 90 studies (41%, Table 3) reviewed contained immunoassay results. It is also possible to obtain indirect information about immunogenicity from efficacy studies, even in the absence of immunoassay results. If NABs are elicited after administration of a therapeutic protein, the resulting antibody-drug complex is often physiologically inactive or may be cleared more rapidly resulting in diminished efficacy. This lack of efficacy was not observed in the 53 studies that lacked immunoassay results. Little evidence for increased immunogenicity and/or associated safety issues were found during this review, consistent with recently published expert opinions of health authority regulators and physician consensus recommendations [34, 35].
The experience with the reference medicine is the best source of data as to what immunogenicity may be expected for a biosimilar. These data have been collected over at least a decade from pharmacovigilance monitoring of the reference medicine, before approval of any corresponding biosimilar. The most important concern related to immunogenicity for all biologics are NABs that lead to loss of efficacy. While the absolute ADA values cannot be compared from one study to the next given the different methodologies, patient populations and medicines assessed, it is possible to draw comparisons within individual studies, and conclusions from each study can be compared. This was the approach utilized in this review.
An efficient immune response against therapeutic proteins depends on the participation of T-cells. T-cell activation, however, only occurs if a portion of the therapeutic protein is presented to them as a relatively small, linear peptide. Given that the primary structures of biosimilars and reference medicines are identical, it follows that peptide epitopes presented to T-cells of the immune system will be the same for both biosimilars and their corresponding reference medicines. Any antibodies developed against the protein backbone of the reference can be expected to cross react with the corresponding biosimilar, and vice versa. In case of differences in post-translational modifications, it is possible that biosimilars may contain unique epitopes that are not presented by reference medicines and that are T-cell independent. It is therefore important to study cross-reactivity in immunogenicity of biosimilars and reference medicines. Two studies were published that evaluated the cross-reactivity of antibodies to reference infliximab and biosimilar infliximab [36, 37]. Both studies concluded that patients who lose responsiveness to reference infliximab due to ADAs will also respond poorly to biosimilar infliximab. Not only is the infliximab biosimilar structurally, functionally, and clinically similar to the reference infliximab, but these studies confirm that the same is true for the immune responses elicited.
This review of the published switching studies also did not show loss of efficacy related to switching from reference medicines to biosimilars. Similarly, there were no new AEs detected in any of the published switching studies that were not already known from studies of the reference medicine. The incidence of safety events in all reported studies was the same before and after the switching event. There is an oft-cited example of increased pure-red cell aplasia (PRCA) that was observed after a major change of a formulation to a marketed erythropoietin [38]. The change included the elimination of the human serum albumin stabilizer from the drug product, which subsequently led to high levels of impurities leaching from the primary packaging. It was established that the PRCA events were caused by an increase in inherent immunogenicity of the medicine. The manufacturer introduced a number of product improvements that restored low immunogenicity [39, 40].
A recent literature case report presented data on an individual that developed serum sickness-like disease after switching from reference medicine to biosimilar infliximab [41]. Serum sickness is a hypersensitivity reaction, commonly occurring a few days to 2 weeks after exposure to a foreign protein or serum component. While this case may be temporally plausible, serum sickness-like disease is already reported as a potential AE that may be encountered after extended treatment with infliximab [42]. Although we lack definitive evidence for this case report to ascertain whether or not this case was triggered by the switch, this case report highlights the need to continue to be vigilant in monitoring the safety of all protein-based therapies.
Kang et al. [13] and Yazici et al. [15] report loss of efficacy or high drop-out rates after switching from reference medicine to biosimilar infliximab. Certainly all such reports need to be considered carefully to determine if there is an emerging signal or pattern. But we note that the results of Kang et al. and Yazici et al. were not replicated in other studies of switching from reference infliximab to biosimilar infliximab. Given that the vast majority of other studies, irrespective of design or size, do not show immunogenicity or signs of intolerance after switching from reference biologic to biosimilar, it is most likely that these two reports are outliers.
Most studies only evaluated a single switch from reference biologic to a biosimilar. Suggestions have been made that there may be an increased safety risk if patients are switched back and forth multiple times between reference biologic and biosimilar. But while the field of biosimilars is new, biologic drugs have been used by patients for decades. Patients have already been exposed to de facto multiple switches for many originator biologics when product quality attributes changed after one or more manufacturing process modifications were introduced [30, 31]. Additional multiple switching studies with biosimilars will directly address this theoretical concern, but at present there is no evidence available that such switches will impact either safety or efficacy. The US Food and Drug Administration has recently issued a draft guidance describing the data requirements necessary to establish the safety and efficacy of pharmacist initiated switching (known in the USA as “interchangeability”), suggesting that manufacturers conduct a clinical study with multiple switches and that utilizes PK or PD primary endpoints, with efficacy parameters as secondary endpoints and safety provided in a descriptive manner [43]. Three multiple switching studies have been reported, each with a different biosimilar. These studies were completed before the draft guidance was issued and do not meet all the design recommendations of the draft guidance because they used efficacy measures as their primary endpoints, instead of PK or PD measures. Nonetheless, the results of these three multiple switching studies do not reveal any safety or efficacy concerns with multiple switches.
Inotai et al. [44] utilized a different approach to assess the concerns expressed in the literature about switching from reference medicines to biosimilars. They identified all articles in which concerns were raised, and then sought to identify evidence supporting these assertions. They concluded that while the hypothetical risk is valid, the assertions have not been supported by “solid scientific evidence.” The extensive review of existing literature as reported here supports this conclusion as well.
4.1 Limitations
An important limitation of this literature review was the variability in methods used by individual studies. By design, this review was designed to be inclusive and as such did not censor reports based on completeness of method description or the rigor in which the studies were executed. Abstracts were included when they were the sole source of data for a given study, but abstracts often lack sufficient details to allow for a full assessment of methodology used. Furthermore, evaluation of the quality of the methods of an individual study is itself subjective; and it was not possible to create non-subjective inclusion/exclusion criteria based on the descriptions of study methods provided in the publications.
The majority of the studies were descriptive in nature and were not powered or designed to detect switch-related differences. As a result, it was not possible to pool the studies in a meta-analysis for either safety or efficacy endpoints.
The small size or the lack of statistical analyses conducted for some studies are limitations that might lead some to dismiss results from those studies; but we elected to include them because descriptive analyses are still a valid comparative tool.
5 Conclusions
There is a large body of published evidence for biologic medicines evaluating the impact of switching from reference medicines to biosimilars that assesses immunogenicity, efficacy, and safety. As of 30 June 2017, 90 switching studies were published with varying study designs, endpoints, and medicines. These included studies in 17 disease indications, in patients and in healthy volunteers, comprising a total of 14,225 subjects. While there are limitations to some of the individual studies, the cumulative results of these published data do not show significant differences in ADAs or NABs after switching compared to subjects that were not switched. There were also no reported increases in treatment-related safety events, including loss of efficacy, that were related to the act of switching from reference medicines to corresponding biosimilars. Thus, the extensive data collected to date suggest that the act of switching from a reference medicine to a biosimilar is not inherently dangerous, and that patients, healthcare professionals, and the public should not assume that it is problematic. As with all biologics, continued pharmacovigilance is important to monitor for rare safety events and for unexpected changes in efficacy or safety profiles that may occur after a manufacturing process change. Furthermore, continued and thorough pharmacovigilance for all biologics should increase confidence of patients, healthcare professionals, and the public in biosimilars, leading to increased acceptance of these safe and effective medicines.
References
US FDA. Information on Biosimilars. 2017. https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/. Accessed 01 Sept 2017.
European Medicines Agency. Questions and answers on biosimilar medicines: EMA2012 27 Sept 2012.
US Public Health Service Act. Section 7002(b)(3) of the Affordable Care Act, adding section 351(i)(2).
Comment from Biosimilars Medicines Group, A Medicines for Europe sector group. Docket submission and presentaton to the Oncologic Drugs Advisory Committee meeting of 13 July 2017. https://www.regulations.gov/document?D=FDA-2017-N-2732-0006. Accesed 21 Nov 2017.
ICH Harmonised Tripartite Quality Guideline 5E (Q5E). Comparability of biotechnological/biological products subject to changes in their manufacturing process. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf; and http://www.ich.org/products/guidelines/quality/quality-single/article/comparabilityof-biotechnologicalbiological. Accessed 30 Aug 2017.
Moots R, Azevedo V, Coindreau JL, Dorner T, Mahgoub E, Mysler E, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19(6):37. https://doi.org/10.1007/s11926-017-0658-4.
Chingcuanco F, Segal JB, Kim SC, Alexander GC. Bioequivalence of biosimilar tumor necrosis factor-alpha inhibitors compared with their reference biologics: a systematic review. Ann Intern Med. 2016;165(8):565–74. https://doi.org/10.7326/M16-0428.
Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473–85. https://doi.org/10.1517/14712598.2012.711308.
National Center for Biotechnology Innovation. MeSH—biosimilar pharmaceutcals. https://www.ncbi.nlm.nih.gov/mesh/68059451. Accessed 30 Jan 2018.
Jalali S, Wohlin C, editors. Systematic literature studies: database searches vs. backward snowballing. Proceedings of the ACM-IEEE international symposium on Empirical software engineering and measurement; 2012: ACM.
Armstrong R, Jackson N, Doyle J, Waters E, Howes F. It’s in your hands: the value of handsearching in conducting systematic reviews of public health interventions. J Public Health (Oxf). 2005;27(4):388–91. https://doi.org/10.1093/pubmed/fdi056.
Higgins J, Lasserson T, Chandler J, Tovey D, Churchill R. Standards for the conduct and reporting of new cochrane intervention reviews, reporting of protocols and the planning, conduct and reporting of updates. Methodological expectations of cochrane intervention reviews (version 1.04, Oct 2017). http://community.cochrane.org/mecir-manual/standards-conduct-new-cochrane-intervention-reviews-c1-c75/performing-review-c24-75/selecting-studies-include-review-c39-42. Accessed 17 Nov 2017.
Kang YS, Moon HH, Lee SE, Lim YJ, Kang HW. Clinical experience of the use of CT-P13, a biosimilar to infliximab in patients with inflammatory bowel disease: a case series. Dig Dis Sci. 2015;60(4):951–6. https://doi.org/10.1007/s10620-014-3392-z.
Jorgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304–16. https://doi.org/10.1016/S0140-6736(17)30068-5.
Yazici Y, Xie L, Ogbomo A, Parenti D, Goyal K, Teeple A, et al. A descriptive analysis of real-world treatment patterns in a Turkish rheumatology population that continued innovator infliximab (Remicade) therapy or switched to biosimilar infliximab. Arthritis Rheumatol. 2016;68(Supp 10): abstract 2240.
ACR-endorsed Criteria for Rheumatic Diseases. American College of Rheumatology. https://www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria. Accessed 21 Nov 2017.
Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Urena S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346–54. https://doi.org/10.1136/annrheumdis-2015-208783.
Yoo DH, Racewicz A, Brzezicki J, Yatsyshyn R, Arteaga ET, Baranauskaite A, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2016;18:82. https://doi.org/10.1186/s13075-016-0981-6.
Choe JY, Prodanovic N, Niebrzydowski J, Staykov I, Dokoupilova E, Baranauskaite A, et al. A randomised, double-blind, phase III study comparing SB2, an infliximab biosimilar, to the infliximab reference product Remicade in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):58–64. https://doi.org/10.1136/annrheumdis-2015-207764.
Glintborg B, Sorensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76(8):1426–31. https://doi.org/10.1136/annrheumdis-2016-210742.
Fiorino G, Manetti N, Armuzzi A, Orlando A, Variola A, Bonovas S, et al. The PROSIT-BIO cohort: a prospective observational study of patients with inflammatory bowel disease treated with infliximab biosimilar. Inflamm Bowel Dis. 2017;23(2):233–43. https://doi.org/10.1097/MIB.0000000000000995.
Rodgers M, Epstein D, Bojke L. Etanercept, infliximab and adalimumab for the treatment of psoriatic artritis: a systematic review and economic evaluation. Appendix 18: estimation of psoriasis area and severity index for treatment responders in the decision model. (2011) NIHR Journals Library, Southampton UK.
Blauvelt A, Puig L, Chimenti S, Vender R, Rajagopalan M, Romiti R, et al. Biosimilars for psoriasis: clinical studies to determine similarity. Br J Dermatol. 2017;177(1):23–33. https://doi.org/10.1111/bjd.15067.
Griffiths CEM, Thaci D, Gerdes S, Arenberger P, Pulka G, Kingo K, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176(4):928–38. https://doi.org/10.1111/bjd.15152.
Papp K, Bachelez H, Costanzo A, Foley P, Gooderham M, Kaur P, et al. Clinical similarity of biosimilar ABP 501 to adalimumab in the treatment of patients with moderate to severe plaque psoriasis: A randomized, double-blind, multicenter, phase III study. J Am Acad Dermatol. 2017;76(6):1093–102. https://doi.org/10.1016/j.jaad.2016.12.014.
Carlsson G, Ahlin A, Dahllof G, Elinder G, Henter JI, Palmblad J. Efficacy and safety of two different rG-CSF preparations in the treatment of patients with severe congenital neutropenia. Br J Haematol. 2004;126(1):127–32. https://doi.org/10.1111/j.1365-2141.2004.05008.x.
Krendyukov A, Harbeck N, Gascon P, Gattu S, Li Y, Blackwell KL. Safety and efficacy of alternating treatment with EP2006, a filgrastim biosimilar, and reference filgrastim for the prevention of severe neutropenia, in patients with breast cancer receiving myelosuppressive chemotherapy. J Clin Oncol. 2017;35(15_suppl):10116. https://doi.org/10.1200/jco.2017.35.15_suppl.10116.
Blackwell K, Semiglazov V, Krasnozhon D, Davidenko I, Nelyubina L, Nakov R, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948–53. https://doi.org/10.1093/annonc/mdv281.
Blauvelt A, Lacour J-P, Fowler JF, Schuck E, Jauch-Lembach J, Balfour A et al. A phase III confirmatory study comparing GP2017 with reference adalimumab in patients with moderate-to-severe chronic plaque psoriasis: 51 week results from the ADACCESS study. European Academy of Dermatology and Venereology Annual Congress; 13–17 Sept 2017; Geneva, Switzerland: EADV-2017; 2017.
Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72:315–8.
Vezer B, Buzas Z, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016;32(5):829–34. https://doi.org/10.1185/03007995.2016.1145579.
Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99–109. https://doi.org/10.3109/1547691X.2013.821564.
Murdaca G, Spano F, Contatore M, Guastalla A, Penza E, Magnani O, et al. Immunogenciity of infliximab and adalimumab: what is its role in hypersensitivity and modulation of therapeutic efficacy and safety? Exp Opin Drug Saf. 2016;15(1):43–52. https://doi.org/10.1517/14740338.2016.1112375.
Kay J, Schoels MM, Dorner T, Emery P, Kvien TK, Smolen JS, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2017. https://doi.org/10.1136/annrheumdis-2017-211937.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31(2):83–91. https://doi.org/10.1007/s40259-017-0210-0.
Ben-Horin S, Yavzori M, Benhar I, Fudim E, Picard O, Ungar B, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65(7):1132–8. https://doi.org/10.1136/gutjnl-2015-309290.
Ruiz-Arguello MB, Maguregui A, Ruiz Del Agua A, Pascual-Salcedo D, Martinez-Feito A, Jurado T, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75(9):1693–6. https://doi.org/10.1136/annrheumdis-2015-208684.
Casadevall N, Eckardt KU, Rossert J. Epoetin-induced autoimmune pure red cell aplasia. J Am Soc Nephrol. 2005;16(Suppl 1):S67–9.
Boven K, Knight J, Bader F, Rossert J, Eckardt KU, Casadevall N. Epoetin-associated pure red cell aplasia in patients with chronic kidney disease: solving the mystery. Nephrol Dial Transplant. 2005;20(Suppl 3):33–40. https://doi.org/10.1093/ndt/gfh1072.
Schellekens H, Jiskoot W. Erythropoietin-associated PRCA: still an unsolved mystery. J Immunotoxicol. 2006;3(3):123–30. https://doi.org/10.1080/15476910600845567.
Scherlinger M, Schaeverbeke T, Truchetet M-E. Serum sickness-like disease after switching to biosimilar infliximab. Rheumatology. 2017. https://doi.org/10.1093/rheumatology/kex268.
Hanauer SB, Feagan BG. Lichtenstein GR Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–9.
US Food and Drug Administration. Guidance for Industry. Considerations in demonstrating interchangeability with a reference product (draft, January 2017) https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed 20 Nov 2017.
Inotai A, Prins CPJ, Csanadi M, Vitezic D, Codreanu C, Kalo Z. Is there a reason for concern or is it just hype?—A systematic literature review of the clinical consequences of switching from originator biologics to biosimilars. Expert Opin Biol Ther. 2017;17(8):915–26. https://doi.org/10.1080/14712598.2017.1341486.
Acknowledgements
We would like to thank Martin Schiestl and Ines Brueckmann for their helpful and insightful comments throughout the planning, research, and writing of this paper.
Funding
No funding was received by the authors for writing this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
HPC is an employee of Sandoz Inc. AB served as a scientific advisor and/or clinical study investigator for AbbVie, Aclaris, Allergan, Almirall, Amgen, Boehringer Ingelheim, Celgene, Dermavant, Dermira, Inc., Eli Lilly and Company, Genentech/Roche, GlaxoSmithKline, Janssen, Leo, Merck Sharp & Dohme, Novartis, Pfizer, Purdue Pharma, Regeneron, Sandoz, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, UCB Pharma, Valeant, and Vidac, and as a paid speaker for Eli Lilly and Company, Janssen, Regeneron, and Sanofi Genzyme. RMR is an employee of McKesson Specialty Health/ The US Oncology Network – The Woodlands, TX, and has served on advisory boards relevant to biosimilars at Amgen, Coherus, EMD Serono (Fresenius) and Pfizer. SD served as a speaker, consultant, and/or advisory board member for AbbVie, Allergan, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Ferring, Hospira, Johnson and Johnson, Janssen, EMD-Serono (Fresenius), MSD, Sandoz, Takeda, Mundipharma, Pfizer Inc, Tigenix, UCB Pharma, and Vifor. SBG is an employee of Novartis Ltd. GW is an employee of Avalere Health.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cohen, H.P., Blauvelt, A., Rifkin, R.M. et al. Switching Reference Medicines to Biosimilars: A Systematic Literature Review of Clinical Outcomes. Drugs 78, 463–478 (2018). https://doi.org/10.1007/s40265-018-0881-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0881-y