
Abstract
Introduction
Although the human epidermal growth factor receptor 2 (HER2) blocker trastuzumab is generally well tolerated, cardiotoxicity can be an important therapeutic limitation.
Objective
In this prespecified analysis, we compared the cardiac safety of the trastuzumab biosimilar ABP 980 (KANJINTI™) and the trastuzumab reference product (RP; Herceptin®) in the phase III LILAC study (ClinicalTrials.gov identifier NCT01901146).
Methods
In the neoadjuvant phase of LILAC, after run-in chemotherapy, 725 patients were randomized 1:1 to ABP 980 (n = 364) or trastuzumab RP (n = 361) plus paclitaxel (every 3 weeks [Q3W] or every week [QW]) for four cycles. After surgery, patients continued treatment Q3W for up to 1 year; ABP 980-treated patients continued ABP 980 (ABP 980/ABP 980; n = 364), and trastuzumab RP-treated patients either continued on the RP (trastuzumab RP/trastuzumab RP; n = 190) or switched to ABP 980 (trastuzumab RP/ABP 980; n = 171). Cardiac safety was monitored by computerized 12-lead electrocardiogram, and left ventricular ejection fraction (LVEF) was assessed by two-dimensional (2D) echocardiogram. LVEF decline was defined as LVEF value decrease from study baseline by ≥ 10 percentage points and to < 50%.
Results
Over the entire study, 22 (3.1%) patients had protocol-defined LVEF decline; no meaningful between-group differences were observed (ABP 980/ABP 980: 2.8%; trastuzumab RP/trastuzumab RP: 3.3%; trastuzumab RP/ABP 980: 3.5%). The incidence of cardiac adverse events was low and comparable in the treatment groups. One grade 3 cardiac failure event reported in the trastuzumab RP/ABP 980 arm, and another in the trastuzumab RP/trastuzumab RP arm, were coincident with LVEF decline. One patient discontinued the investigational product during the adjuvant phase because of cardiac failure.
Conclusion
These prespecified analyses confirm the tolerability of ABP 980 and demonstrate clinical similarity of ABP 980 and trastuzumab RP with respect to cardiac safety. No new cardiac safety signals were observed whether patients were receiving ABP 980 or switched from the RP to ABP 980.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cardiotoxicity is an important therapeutic limitation of the human epidermal growth factor receptor 2 (HER2) blocker trastuzumab. |
ABP 980 is a biosimilar to trastuzumab (Herceptin®) reference product (RP). |
The cardiac safety of ABP 980 was compared with its RP in the phase III LILAC study, and the incidence of cardiac adverse events was found to be low and comparable in the treatment groups, with no new cardiac safety signals observed with ABP 980. |
1 Introduction
ABP 980 is a biosimilar to Herceptin® (trastuzumab; Roche Registration GmbH, Germany; Genentech, San Francisco, USA). Herceptin® is approved for use in the United States (US), the European Union (EU), Japan, and much of the rest of the world for the treatment of metastatic breast cancer, early breast cancer, and metastatic gastric cancer, and is the standard of care for subjects with human epidermal growth factor receptor 2 (HER2)-overexpressing breast cancer [1, 2]. In the recent phase III LILAC trial (ClinicalTrials.gov identifier NCT01901146), ABP 980 was shown to be similar to the trastuzumab reference product (RP) with respect to efficacy, safety, and immunogenicity in women with HER2-positive early breast cancer, with no clinically meaningful difference between the two products [3]. The LILAC study found that the frequency, type, and severity of adverse events (AEs), including cardiac events, were comparable between treatment arms and were consistent with the known safety profile of trastuzumab [1, 3, 4]. Based in part on these data, and the totality of evidence generated during development, ABP 980 (KANJINTI™ [trastuzumab], Amgen Europe B.V., The Netherlands) was approved by the European Medicines Agency in May 2018 for the same indications as Herceptin®, including the treatment of HER2-positive metastatic breast cancer, HER2-positive early breast cancer, and HER2-positive metastatic adenocarcinoma of the stomach or gastroesophageal junction [5]. Since then, the US FDA has also approved ABP 980 (KANJINTI™ [trastuzumab-anns], Amgen Inc., Thousand Oaks, CA, USA) for all approved indications of the RP [6].
While trastuzumab is generally well-tolerated, cardiac toxicity remains a rare but serious concern and may limit the therapeutic potential of trastuzumab in a subset of patients [7]. HER2 is known to play an important role in cardiac development and may also play a physiologic role in normal cardiac function [7, 8]. The mechanism of cardiac toxicity is not entirely understood but studies suggest that it may involve HER2 blockade. Trastuzumab-induced cardiotoxicity may result from decreased HER2 signaling required for cardiac contractility, interference with cardiomyocyte survival signals, and compromised myocyte stress response [7, 8].
Left ventricular systolic dysfunction is the most concerning cardiac toxicity associated with trastuzumab, particularly when it is administered in combination with an anthracycline [7]. Adjuvant clinical trials of trastuzumab have reported severe heart failure in 1.44–4% of patients; the overall risk ratio (RR) of high-grade heart failure was reported to be 3.04-fold higher with trastuzumab than without [9, 10]. Moreover, up to 19% of patients were reported to have developed a significant decline in left ventricular ejection fraction (LVEF) [9, 11]. However, the TRYPHAENA study, which assessed the cardiac safety of trastuzumab and pertuzumab with chemotherapy during the neoadjuvant treatment of 225 women with HER2-positive early breast cancer, showed a low incidence of symptomatic left ventricular systolic dysfunction [12, 13].
In this study, we examine in greater detail the cardiac safety of ABP 980 compared with trastuzumab RP in the LILAC study.
2 Materials and Methods
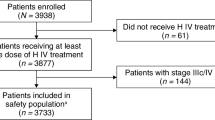
The study design is shown in Fig. 1 and has been described in detail previously [3]. Briefly, this randomized, multicenter, double-blind, active-controlled equivalence trial compared ABP 980 with trastuzumab RP in adult women with HER2-positive early breast cancer. Patients included women ≥ 18 years of age with histologically confirmed invasive breast cancer and with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, were planned to receive neoadjuvant therapy and subsequent surgical resection of breast tumor and sentinel lymph node dissection (SLND) or axillary lymph node dissection (ALND), had HER2-positive disease (confirmed by a central laboratory before patients were randomized) defined as 3 + overexpression by immunohistochemistry or HER2 amplification by fluorescence in situ hybridization and with known estrogen and progesterone hormone receptor status at study entry, measurable disease in the breast after diagnostic biopsy, defined as the longest diameter ≥ 2.0 cm, and LVEF ≥ 55% by two-dimensional (2D) echocardiogram.

(modified from von Minckwitz et al. [3])
LILAC: trial design
Exclusion criteria included the presence of bilateral breast cancer or known distant metastases; prior treatment including chemotherapy, biologic therapy, radiation, or surgery for primary breast cancer; concomitant active malignancy; and had concomitant active malignancy or a history of malignancy in the past 5 years, except treated basal cell carcinoma of the skin or carcinoma in situ of the cervix.
After a 28-day screening period, patients entered a neoadjuvant treatment period to receive run-in chemotherapy consisting of epirubicin and cyclophosphamide (EC) every 3 weeks (Q3W) for four cycles, followed by surgery within 3–7 weeks of the last dose of investigational product (IP) in the neoadjuvant phase. Once run-in chemotherapy was completed, patients with adequate cardiac function were randomized 1:1 to treatment with ABP 980 (Amgen Inc., Thousand Oaks, CA, USA) or trastuzumab RP (Herceptin®; Genentech, San Francisco, CA, USA) plus paclitaxel Q3W for four cycles; paclitaxel once weekly (QW) was also allowed if consistent with local standard of care. Randomization was stratified according to tumor or T stage (< T4, T4), node status (yes, no), hormone receptor status (estrogen receptor [ER]-negative and/or progesterone receptor [PR]-positive vs. ER-negative and PR-negative), planned paclitaxel dosing schedule (QW, Q3W), and geographic region (Eastern Europe, Western Europe, or other). IP was administered at an initial dose of 8 mg/kg over 90 min, followed by 6 mg/kg intravenous infusions for three cycles.
Surgery (lumpectomy or mastectomy with SLND or ALND) was completed 3–7 weeks after the last dose of IP in the neoadjuvant phase. An adjuvant phase followed surgery, during which patients received ABP 980 or a trastuzumab RP 6 mg/kg intravenous infusion Q3W for up to 1 year starting from the first day of IP administration in the neoadjuvant phase. In the adjuvant phase, patients who initially received ABP 980 during the neoadjuvant phase continued receiving ABP 980 Q3W; patients who initially received trastuzumab RP during the neoadjuvant phase were randomized to either continue receiving trastuzumab Q3W or switched to ABP 980 Q3W (trastuzumab RP/ABP 980 group). Allocation to the neoadjuvant and adjuvant (including the single switch) treatment arm occurred at randomization and was maintained in a blinded manner.
AEs and disease progression or recurrence were assessed at each visit during the neoadjuvant and adjuvant periods.
During the adjuvant phase, the following assessments/evaluations were made: AEs, serious AEs (SAEs), concomitant medications, disease progression or recurrence, and hematology sampling (all cycles); physical examination, vital signs and weight, serum chemistry, anti-drug antibody (ADA) sampling (cycles 5, 9, and 13), and pharmacokinetic sampling. AEs were monitored by investigators or reported by the patients after randomization through 30 days after the last dose of IP.
2.1 Cardiac Monitoring
For cardiac safety monitoring, computerized 12-lead electrocardiogram (ECG) recordings were obtained for heart rate, P, PR, QRS, QT, and QTc intervals. A copy of all ECGs were retained on site and could be collected for central review if required (i.e. in case emerging ECG data necessitated expert evaluation). ECGs could be repeated for quality reasons and additional ECGs could be collected by investigators for safety reasons. Any new clinically relevant abnormal findings were reported as AEs. Patients with clinically relevant cardiovascular adverse effects or AEs (or clinically relevant ECG changes) were withdrawn from study treatment per protocol.
To assess LVEF, a 2D echocardiogram was performed per local standard-of-care guidelines. During the neoadjuvant phase, the 2D echocardiogram was performed at screening, baseline (cycle 1) and at cycle 4 (last visit of the neoadjuvant phase) (Fig. 1). At all indicated visits, the 2D echocardiogram was required to be performed and results obtained prior to IP administration. During the adjuvant phase, 2D echocardiograms were performed at cycles 9 and 13. A 2D echocardiogram was also performed during the end-of-treatment (EOT, cycle 17) and end-of-study (EOS) visits; EOS visits were scheduled 30 days after the last IP, or 1 year from the first IP for subjects who withdrew early but remained on scheduled assessments. Investigators were strongly urged to schedule the LVEF assessment at the same cardiac imaging facility that performed the patient’s baseline LVEF assessment. If IP was discontinued for any reason, protocol-mandated LVEF assessments were obtained.
During IP treatment, if LVEF decreased by ≥ 10% points from the baseline (cycle 1) post-anthracycline/pre-randomization echocardiogram and to < 50%, treatment was suspended and a repeat LVEF assessment was performed within approximately 3 weeks. If LVEF was not improved or if it declined further, IP was to be discontinued. If symptomatic cardiac failure developed during the IP treatment, it was treated according to local standard of care. The IP was required to be discontinued in subjects who developed clinically significant heart failure. Based on the known safety profile of trastuzumab, treatment-emergent cardiac failure was a prespecified event of interest (EOI) that was monitored closely during the study.
3 Results
Key demographics and baseline characteristics are summarized in Table 1. For a complete list of demographic and baseline characteristics see von Minckwitz et al. [3]. In general, the treatment groups were well balanced. At baseline, by neoadjuvant treatment, the mean LVEF was 64.67% (standard deviation [SD] 5.143) and 64.60% (SD 4.837) in the ABP 980 and trastuzumab arms, respectively. Cardiac findings at screening by neoadjuvant and neoadjuvant/adjuvant treatment for resolved and unresolved findings are summarized in Tables 2 and 3, respectively.
3.1 Left Ventricular Ejection Fraction Decline
On average, LVEF did not change over the course of the study and was comparable across the three treatment groups (Fig. 2). The number of patients experiencing LVEF decline by ≥10% and to < 50% was low and comparable across treatment groups (Table 4). Across the entire study, a total of 22 patients (3.1%) experienced LVEF decline by ≥10% and to < 50%; the differences between treatment arms (ABP 980/ABP 980: 10 [2.8%]; trastuzumab RP/trastuzumab RP: 6 [3.3%]; trastuzumab RP/ABP 980: 6 [3.5%]) were not clinically meaningful. There were no meaningful differences between treatment arms in LVEF changes from baseline based on the lowest value reported for LVEF by neoadjuvant and adjuvant treatment. Overall, during the neoadjuvant phase, the incidence of LVEF decline was reported in 1 (0.3%) patient in the ABP 980 arm and 3 (0.9%) patients in the trastuzumab RP arm. The incidence of LVEF decline was higher in the adjuvant phase, but was similar between the treatment arms (ABP 980/ABP 980: 10 [2.9%]; trastuzumab RP/trastuzumab RP: 3 [1.8%]; trastuzumab RP/ABP 980: 6 [3.6%]).

LVEF: summary by cycle. Baseline is defined as the last non-missing assessment taken prior to the first dose of study IP. Cycle 4 = visit 9; cycle 9 = visit 14; cycle 13 = visit 18. EOS visits were scheduled 30 days after the last IP, or 1 year from the first IP for subjects who withdrew early but remained on scheduled assessments. Results from unscheduled visits are included in these summaries. LVEF left ventricular ejection fraction, IP investigational product, EOS end of study
Assessment of onset time of LVEF decline (by ≥ 10% and to < 50%) during the approximately 1 year of trastuzumab or ABP 980 treatment duration showed no definite patterns. Of the 22 events, 6 occurred before cycle 9, 6 between cycles 9 and 13, and 10 by EOT (cycle 13) or EOS (Table 5).
The majority of patients with LVEF decline had a prior history of cardiovascular and metabolic disorders, including hypertension, arrhythmia, atrial fibrillation, arterial hypertension, venous insufficiency, hypercholesterolemia, and diabetes (Table 5). At screening, one patient had a slight delay of ventricular relaxation, and another had mild diastolic dysfunction; however, their LVEF values were normal at study initiation (Table 5).
The majority of the LVEF declines were asymptomatic. There were three cardiac failure events (a prespecified EOI; discussed further in the following section), and three cases of ventricular hypokinesia that were coincident with LVEF decline by ≥ 10% points and to < 50% compared with baseline (Table 5).
3.2 Cardiac Failure
The incidence of cardiac disorder AEs is shown in Table 6. The incidence of any cardiac disorder was low across treatment groups (ABP 980/ABP 980: 35 [9.6%]; trastuzumab RP/trastuzumab RP: 14 [7.4%]; trastuzumab RP/ABP 980: 20 [11.7%]).
Overall, the incidence of cardiac failure was comparable in the ABP 980/ABP 980 (8 [2.2%]), trastuzumab RP/trastuzumab RP (1 [0.5%]), and trastuzumab RP/ABP 980 (2 [1.2%]) groups. Most cardiac failure EOIs reported over the entire study were grade 1 (n = 7) or grade 2 (n = 3); none were grades 4 or 5. None of the cardiac failure EOIs were SAEs. All 11 subjects with cardiac failure EOIs were 50 years of age or older. Of the eight subjects with cardiac failure EOIs in the ABP 980/ABP 980 treatment arm, four had a relevant ongoing medical history of cardiac disease that may have contributed to cardiac failure EOIs. All 11 subjects who experienced cardiac failure EOIs underwent surgery and entered the adjuvant phase; one subject did not complete the planned IP dose and discontinued due to this AE; all others completed all planned doses of IP. In one subject (trastuzumab RP/ABP 980), the LVEF decline and cardiac failure AE had the same onset date, and the AE was considered possibly related. In two subjects, the cardiac failure AE was considered unlikely related: one subject (trastuzumab RP/trastuzumab RP) had cardiac failure 6 months after the LVEF decline, and one subject (ABP 980/ABP 980) had cardiac failure 3 months before LVEF decline. Of the 11 subjects, one subject in the ABP 980/ABP 980 arm had improved LVEF function at a subsequent visit (overall assessment of ‘normal’ following the report of grade 2 chronic cardiac failure; however, the subject experienced LVEF decline to < 50% at the EOS visit).
During the neoadjuvant phase, seven patients experienced any cardiac failure AE (ABP 980: 6 [1.6%]; trastuzumab RP: 1 [0.3%]), and none experienced cardiac failure coincident with LVEF decline by ≥ 10% points and to < 50% compared with baseline. Cardiac failure events were grade 1 or 2 and patients completed all planned doses of IP, indicating resolution or no worsening of the cardiac failure event.
During the adjuvant phase, 2 (0.6%), 1 (0.6%), and 1 (0.6%) patients in the ABP 980/ABP 980, trastuzumab RP/trastuzumab RP, and trastuzumab RP/ABP 980 groups, respectively, had at least one cardiac failure event. All cardiac failure events in the ABP 980/ABP 980 and trastuzumab RP/trastuzumab RP treatment groups were grade 1 or 2. One patient in the trastuzumab RP/ABP 980 arm (0.6%) had a cardiac failure event of grade 3 severity; all other cardiac failure events in this treatment group were grade 1 or 2. The grade 3 cardiac failure event was coincident with LVEF decline by ≥ 10% points and to < 50% compared with baseline. This cardiac failure event occurred 371 days after study drug initiation, was considered related to the study drug, and the patient recovered/resolved with sequelae; the 79-year-old patient had no relevant cardiovascular medical history. One patient discontinued IP during the adjuvant phase because of cardiac failure.
4 Discussion
The results of the phase III LILAC study previously demonstrated that ABP 980 is similar to trastuzumab RP with respect to efficacy, safety, and immunogenicity in women with HER2-positive early breast cancer. Here, we extend these results by presenting the outcomes of a prespecified cardiac safety analysis of ABP 980 in that study. Although the mechanism is still not fully understood, cardiotoxicity is the most important limitation in the application of trastuzumab. Thus, cardiac safety is one of the most important parameters to carefully monitor when assessing the safety of a trastuzumab biosimilar.
Our results demonstrate a low incidence of cardiotoxicity following long-term (up to 1 year) treatment with the trastuzumab biosimilar ABP 980 in combination with chemotherapy. No correlation was observed between subjects with an LVEF decline of ≥ 10 percentage points and to < 50% and subjects who experienced a cardiac failure EOI. There were no clinically meaningful differences in the incidence of LVEF decline between treatment arms in the adjuvant phase or over the entire study.
The limitation of the current analysis is that the study was not statistically powered to detect differences in cardiac parameters among the treatment groups, and that the small number of cardiovascular events noted during the study preclude more robust analyses.
Overall, the incidence of cardiac failure frequency in the LILAC study was within the expected range based on the label and published data for trastuzumab RP [1, 4]. Moreover, our analysis demonstrated comparable cardiac safety in patients who were switched from trastuzumab RP to ABP 980 following surgery compared with patients who were maintained on treatment with either ABP 980 or trastuzumab RP. This finding has important implications for clinicians who may consider switching patients from trastuzumab RP to ABP 980.
5 Conclusions
The results of this prespecified analysis of the LILAC study confirm the tolerability of ABP 980 and further demonstrate acceptable cardiac safety, which is a particular concern with trastuzumab therapy. No new or unexpected cardiac safety signals were observed with ABP 980 compared with trastuzumab RP, whether patients were started on ABP 980 or were switched from trastuzumab RP to ABP 980.
References
Genentech. Herceptin® (trastuzumab) prescribing information. South San Francisco: Genentech; 2018.
Roche Pharma AG. Herceptin® (trastuzumab) summary of product characteristics. Grenzach-Wyhlen: Roche Pharma AG; 2018.
von Minckwitz G, Colleoni M, Kolberg HC, Morales S, Santi P, Tomasevic Z, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987–98.
Burki TK. Trastuzumab cardiotoxicity in early-stage breast cancer. Lancet Oncol. 2016;17(6):e226.
Amgen NV. Kanjinti (trastuzumab) summary of product characteristics. Diegem: Amgen NV; 2018.
Amgen Inc. Kanjinti (trastuzumab anns) for injection, for intravenous use. Thousand Oaks: Amgen Inc.; 2019.
Chien AJ, Rugo HS. The cardiac safety of trastuzumab in the treatment of breast cancer. Expert Opin Drug Saf. 2010;9(2):335–46.
An J, Sheikh MS. Toxicology of trastuzumab: an insight into mechanisms of cardiotoxicity. Curr Cancer Drug Targets. 2019;19(5):400–7.
Romond EH, Jeong JH, Rastogi P, Swain SM, Geyer CE Jr, Ewer MS, et al. Seven-year follow-up assessment of cardiac function in NSABP B-31, a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel (ACP) with ACP plus trastuzumab as adjuvant therapy for patients with node-positive, human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2012;30(31):3792–9.
Long HD, Lin YE, Zhang JJ, Zhong WZ, Zheng RN. Risk of congestive heart failure in early breast cancer patients undergoing adjuvant treatment with trastuzumab: a meta-analysis. Oncologist. 2016;21(5):547–54.
Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–83.
Schneeweiss A, Chia S, Hickish T, Harvey V, Eniu A, Hegg R, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol. 2013;24(9):2278–84.
Schneeweiss A, Chia S, Hickish T, Harvey V, Eniu A, Waldron-Lynch M, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer. 2018;89:27–35.
Acknowledgements
The authors thank the patients, investigators, and all those who participated in the study. Medical writing support was funded by Amgen Inc. and was provided by Pasquale Iannuzzelli, Ph.D. (Innovation Communications Group, New York, NY, USA) under the direction of Monica Ramchandani, Ph.D. (Amgen Inc., Thousand Oaks, CA, USA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Hans-Christian Kolberg received honoraria from Carl Zeiss Meditec, TEVA, Theraclion, Novartis, Amgen, Astra Zeneca, Pfizer, Janssen-Cilag, GSK, LIV Pharma, Roche and Genomic Health, and non-financial support from Carl Zeiss Meditec, Novartis, Pfizer, Amgen, Roche, LIV Pharma, Tesaro, Daiichi Sankyo, and Genomic Health. Marco Colleoni is a consultant for AstraZeneca, Celldex, Novartis, OBI Pharma, Pfizer, Pierre Fabre, and Puma Biotechnology, has received honoraria from Novartis, and is also an investigator on the ABP 980 LILAC Study. Georgia Savva Demetriou is a consultant for Astra Zeneca and Celgene, and was on speakers’ bureaus for Merck and Serono. Patricia Santi was an investigator on the ABP 980 LILAC Study, but has no other conflicts to declare. Hans Tesch received honoraria from Novartis, Roche, Celgene, Teva, and Pfizer, and travel support from Roche, Celgene, and Pfizer. Yasuhiro Fujiwara received lecture fees from AstraZeneca KK, Daiichi-Sankyo, Taiho, Chugai, Novartis Pharma KK, Bristol-Myers KK, SRL, and Santen Pharmaceutical. Zorica Tomasevic is a consultant for and received honoraria from Roche, Pfizer, and Novartis, has served on the speakers’ bureau for Pfizer, and has received travel support from Roche. Vladimir Hanes is an employee and stockholder of Amgen.
Funding
This study was funded by Amgen, Inc., Thousand Oaks, CA, USA.
Ethical Approval
The protocol was reviewed and approved by the relevant independent Ethics Committees for each center. All patients provided written informed consent. This study was conducted in accordance with the terms of the Declaration of Helsinki, Good Clinical Practice guidelines, and all applicable regulatory requirements.
Data Availability
There is a plan to share data. This may include de-identified individual patient data for variables necessary to address the specific research question in an approved data-sharing request, as well as related data dictionaries, study protocol, statistical analysis plan, informed consent form, and/or clinical study report. Data-sharing requests relating to data in this article will be considered after the publication date and (1) this product and indication (or other new use) have been granted marketing authorization in both the US and Europe, or (2) clinical development discontinues and the data will not be submitted to regulatory authorities. There is no end date for eligibility to submit a data-sharing request for these data. Qualified researchers may submit a request containing the research objectives, the Amgen product(s) and Amgen study/studies in scope, endpoints/outcomes of interest, statistical analysis plan, data requirements, publication plan, and qualifications of the researcher(s). In general, Amgen does not grant external requests for individual patient data for the purpose of re-evaluating safety and efficacy issues already addressed in the product labeling. A committee of internal advisors reviews requests. If not approved, a Data Sharing Independent Review Panel may arbitrate and make the final decision. Requests that pose a potential conflict of interest or an actual or potential competitive risk may be declined at Amgen’s sole discretion and without further arbitration. Upon approval, information necessary to address the research question will be provided under the terms of a data-sharing agreement. This may include anonymized individual patient data and/or available supporting documents, containing fragments of analysis code, where provided, in analysis specifications. Further details are available at http://www.amgen.com/datasharing.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kolberg, HC., Colleoni, M., Demetriou, G.S. et al. Cardiac Safety of the Trastuzumab Biosimilar ABP 980 in Women with HER2-Positive Early Breast Cancer in the Randomized, Double-Blind, Active-Controlled LILAC Study. Drug Saf 43, 233–242 (2020). https://doi.org/10.1007/s40264-019-00886-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-019-00886-3