
Abstract
Objective
The objective of this randomized, double-blind, placebo-controlled study was to evaluate the efficacy and safety of SHP465 mixed amphetamine salts (MAS) in adults with attention-deficit/hyperactivity disorder (ADHD).
Methods
Eligible adults [aged 18–55 years; meeting the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition ADHD criteria; baseline ADHD Rating Scale with Adult Prompts (ADHD-RS-AP) total scores ≥28] were randomized 1:1:1 to placebo or forced-dose SHP465 MAS (12.5 or 37.5 mg/day) for 4 weeks. The ADHD-RS-AP total score change from baseline to week 4 (primary endpoint) and Clinical Global Impressions-Improvement score at week 4 (key secondary endpoint) were assessed using linear mixed-effects models for repeated measures. Other efficacy endpoints were changes from baseline to week 4 on the ADHD-RS-AP hyperactivity/impulsivity and inattentiveness subscales and the percentage of participants categorized as improved on the dichotomized Clinical Global Impressions-Improvement. Safety and tolerability assessments were treatment-emergent adverse events, vital sign and weight changes, Columbia-Suicide Severity Rating Scale responses, and electrocardiogram results.
Results
Of 369 screened participants, 275 were randomized (placebo, n = 91; 12.5 mg/day of SHP465 MAS, n = 92; 37.5 mg/day of SHP465 MAS, n = 92) and 236 completed the study (placebo, n = 80; 12.5 mg/day of SHP465 MAS, n = 80; 37.5 mg/day of SHP465 MAS, n = 76). Least-squares mean (95% confidence interval) treatment differences at week 4 significantly favored SHP465 MAS over placebo for the ADHD-RS-AP total score change from baseline [12.5 mg/day: −8.1 (−11.7, −4.4), effect size = 0.67; 37.5 mg/day: −13.4 (−17.1, −9.7), effect size = 1.11; both p < 0.001] and Clinical Global Impressions-Improvement score [12.5 mg/day: −0.8 (−1.1, −0.4), effect size = 0.68; 37.5 mg/day: −1.2 (−1.6, −0.9), effect size = 1.11; both p < 0.001]. Treatment differences for the change from baseline at week 4 favored 12.5 and 37.5 mg/day of SHP465 MAS, respectively, over placebo on the ADHD-RS-AP hyperactivity/impulsivity (both nominal p < 0.001; effect size = 0.56 and 0.91) and inattentiveness (both nominal p < 0.001; effect size = 0.70 and 1.19) subscales. At the final on-treatment assessment, the percentage of participants categorized as improved on Clinical Global Impressions-Improvement was higher with both SHP465 MAS doses than with placebo (both nominal p < 0.001). Treatment-emergent adverse events reported (>5%) with SHP465 MAS were decreased appetite, dry mouth, insomnia, headache, anxiety, initial insomnia, irritability, and bruxism. Severe treatment-emergent adverse events and treatment-emergent adverse events leading to discontinuation, respectively, were reported by 8 and 12 participants (placebo, n = 2 and 0; 12.5 mg/day SHP465 MAS, n = 1 and 7; 37.5 mg/day SHP465 MAS, n = 5 and 5). At the final on-treatment assessment, mean ± standard deviation increases from baseline were observed with 12.5 and 37.5 mg/day of SHP465 MAS for pulse (3.3 ± 10.52 and 7.1 ± 11.48 bpm) and blood pressure (systolic 0.2 ± 7.24 and 1.7 ± 9.99 mmHg; diastolic 1.0 ± 7.46 and 2.8 ± 7.90 mmHg) and decreases were observed for weight (−0.97 ± 1.523 and −1.65 ± 2.333 kg), body mass index (−0.33 ± 0.519 and −0.56 ± 0.777 kg/m2), and Fridericia corrected QT interval (−3.0 ± 10.72 and −1.6 ± 13.70 ms). No participant in any treatment group had a positive response for on-study Columbia-Suicide Severity Rating Scale assessments.
Conclusions
SHP465 MAS was superior to placebo in reducing ADHD symptoms, with a safety profile consistent with other long-acting stimulants.
ClinicalTrials.gov Registry Number: NCT02604407.
Similar content being viewed by others

In adults with attention-deficit/hyperactivity disorder (ADHD), 12.5 and 37.5 mg/day of SHP465 mixed amphetamine salts (MAS) produced significantly greater reductions in ADHD symptoms, as measured by the total score on the ADHD Rating Scale with Adult Prompts, than did placebo after 4 weeks of treatment. |
At the end of the study, 37.5 mg/day of SHP465 MAS vs. placebo demonstrated a robust effect size of 1.11 for the primary efficacy endpoint (ADHD Rating Scale with Adult Prompts total score) and the key secondary efficacy endpoint (Clinical Global Impressions-Improvement score), with the effects of 12.5 mg/day of SHP465 MAS being numerically lower than 37.5 mg/day of SHP465 MAS. |
The overall safety and tolerability profile of SHP465 MAS, in terms of treatment-emergent adverse events and vital sign changes, aligned with the known effects of other long-acting amphetamine formulations. |
Taken together with previous findings, SHP465 MAS is a viable treatment for adults with ADHD. |
1 Introduction
Attention-deficit/hyperactivity disorder (ADHD) presents clinically in children and adolescents, and it may persist in a sizeable portion of adults [1,2,3,4]. When pharmacotherapy for adults diagnosed with ADHD is pursued, the initial treatment choice is generally a psychostimulant [5, 6], which is in accordance with US practice recommendations for children and adolescents with ADHD [7]. Although reviews suggest that currently available long-acting psychostimulants have estimated durations of effect ranging from 8 to 14 h [8,9,10], some physicians may augment their patients’ long-acting psychostimulant with another ADHD medication later in the day because the long-acting psychostimulant alone does not provide all-day coverage. In a clinical program practice survey of adults being treated for ADHD, approximately 40% of patients were receiving a combination of short- and long-acting psychostimulants [11].
SHP465 mixed amphetamine salts (SHP465 MAS) is a novel, once-daily, extended-release, single-entity MAS product for oral administration approved in USA for the treatment of ADHD in patients aged 13 years and older. SHP465 MAS was designed with the goal of providing a medication with the potential to reduce the ADHD symptoms throughout the day and into the evening in individuals who require such coverage. It contains equal amounts (by weight) of four salts: dextroamphetamine sulfate and amphetamine sulfate, dextroamphetamine saccharate and amphetamine aspartate monohydrate. This results in a 3:1 mixture of dextro- to levoamphetamine base equivalent. Each capsule contains three types of drug-releasing beads, an immediate-release bead and two different types of delayed-release beads. The first delayed-release bead releases amphetamine at pH 5.5 and the other delayed-release bead releases amphetamine at pH 7.0. In a phase I study, the pharmacokinetic profile of 37.5 mg/day of SHP465 MAS was similar to that of 25 mg/day of extended-release MAS (MAS XR) in the morning supplemented 8 h later by 12.5 mg/day of immediate-release MAS [12], suggesting it may be a viable once-daily treatment for ADHD.
The efficacy, tolerability, and safety of SHP465 MAS in adults with ADHD have been examined in several phase III studies [13,14,15]. In a forced-dose study, the efficacy, tolerability, and safety of 25, 50, and 75 mg/day of SHP465 MAS were examined [15]. In that study, all SHP465 MAS doses produced significantly greater reductions in ADHD symptoms than did placebo, but a clear and clinically meaningful, dose-dependent response between low and high doses of SHP465 MAS was not established [15]. In another study, the efficacy, tolerability, and safety of dose-optimized SHP465 MAS (12.5–75 mg/day) were evaluated; however, owing to the use of dose optimization, relationships among the individual SHP465 MAS doses used in that study (12.5, 25, 37.5, 50, 62.5, and 75 mg/day) were not examined [13]. This forced-dose study specifically evaluated the efficacy, tolerability, and safety of 12.5 and 37.5 mg/day of SHP465 MAS, with the 12.5-mg/day dose being evaluated to further elucidate the lower end of the dose response relationship of SHP465 MAS. The objectives of this study were to evaluate the efficacy, safety, and tolerability of SHP465 MAS vs. placebo in the treatment of adults with ADHD.
2 Materials and Methods
This phase III, randomized, double-blind, parallel-group, placebo-controlled, forced-dose study (ClinicalTrials.gov registry: NCT02604407) was conducted at 43 US sites [participants randomized/enrolled across sites (median 6; range 1–19); no more than 7% of participants enrolled from each study site] in accordance with the International Conference on Harmonisation and Good Clinical Practice and the principles of the Declaration of Helsinki. The study protocol, final approved informed consent document, and all supporting information were submitted to and approved by a central institutional review board (Copernicus Group Independent Review Board, Durham, NC, USA) and by the US Food and Drug Administration as appropriate before study initiation. All participants provided written informed consent before taking part in study procedures.
2.1 Participants
Men and non-pregnant women (aged 18–55 years) meeting the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria for a primary ADHD diagnosis and having ADHD Rating Scale with Adult Prompts (ADHD-RS-AP) total scores ≥28 at baseline were enrolled. Eligible participants had satisfactory medical assessments with no clinically significant abnormalities and they were either not currently on ADHD therapy or they reported that they were not completely satisfied with any aspect, including issues related to efficacy and tolerability, of their current ADHD therapy.
Participants were excluded if they had a comorbid psychiatric diagnosis that was controlled with prohibited medications or uncontrolled and associated with significant symptoms that contraindicated SHP465 MAS treatment or could confound study assessments. Participants were also ineligible if they were considered a suicide risk, had previously made a suicide attempt, or were currently demonstrating active suicidal ideation (those with intermittent passive suicidal ideation could be included based on investigator judgment). Other exclusion criteria included a body mass index (BMI) <18.5 kg/m2 (underweight) or ≥40 kg/m2 (very severely obese) at screening; a history of moderate-to-severe hypertension, average sitting systolic blood pressure (SBP) >139 mmHg, or average sitting diastolic blood pressure (DBP) >89 mmHg; use of combination antihypertensive medications (participants with well-controlled mild hypertension on a single antihypertensive agent could participate); a known history of symptomatic cardiovascular issues or serious cardiac problems; a known family history of sudden cardiac death or ventricular arrhythmia; a clinically significant electrocardiogram; a documented allergy, a hypersensitivity, or an intolerance to amphetamine or any excipient in SHP465 MAS; a failure to respond to an adequate course of amphetamine therapy (to ensure that response to treatment is not artificially reduced by the inclusion of non-responders); a history of suspected substance abuse or dependence disorder (excluding nicotine) based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria; a lifetime history of amphetamine, cocaine, or other stimulant abuse and/or dependence; and participation in a clinical study within 30 days before screening.
Prohibited medications were psychostimulants and amphetamine-like agents, centrally or peripherally acting antihistamines (non-sedating antihistamines were permitted), investigational compounds, clonidine and guanfacine, and herbal preparations. Additional medications that were prohibited because they indicated the potential presence of an exclusionary diagnosis were combination anti-hypertensives (use of a single antihypertensive was permitted if the same dose had been used for at least 3 months before screening), sedatives, anxiolytics, antipsychotics, monoamine oxidase inhibitors, antidepressants, selective noradrenaline reuptake inhibitors, and noradrenaline reuptake inhibitors. Participants requiring or anticipating the need to take medications with central nervous system effects or who were taking monoamine oxidase inhibitors were excluded (stable bronchodilator inhaler use was not exclusionary).
2.2 Treatment
The study included four periods (screening and washout, forced-dose titration, dose maintenance, safety follow-up) (Fig. 1). After the screening and washout period (7–30 days depending on use of prohibited medications at screening), participants were randomized 1:1:1 to 1 of three treatment groups for 4 weeks: 12.5 mg/day of SHP465 MAS, 37.5 mg/day of SHP465 MAS, or matching placebo. Participants were instructed to take their treatment in the morning at approximately 7:00 a.m. (±2 h).

Study design. ET early termination, MAS mixed amphetamine salts, PBO placebo, V visit
Treatment assignments were made by an interactive web response system. Within each study site, unique participant numbers were assigned according to the sequence of presentation for study participation after determination of study eligibility. The packaging of the study drug and the appearance of each treatment were identical across treatment arms. The investigators, the investigators’ staff, and the participants were blinded to the treatment assignment.
During the forced-dose titration period (weeks 1 and 2), participants randomized to SHP465 MAS initiated treatment at 12.5 mg/day during week 1. Participants randomized to 12.5 mg/day of SHP465 MAS maintained this dose during week 2. In participants randomized to 37.5 mg/day of SHP465 MAS, the dose was titrated to 25 mg/day during week 2 and to 37.5 mg/day during week 3. Throughout the forced-dose titration period, dose escalation was dictated by treatment group, and dose changes were not allowed. Participants were discontinued if they experienced unacceptable tolerability. During dose maintenance (weeks 3 and 4), participants were maintained at their randomized dose. The follow-up period was 7 (+2) days from the last dose of the study drug. At the end of the follow-up period, information related to the occurrence of adverse events (AEs) and concomitant treatments was requested via a telephone call.
2.3 Efficacy Endpoints
The primary efficacy endpoint was ADHD-RS-AP total score change from baseline to week 4 in the full analysis set, which included all screened participants assigned a randomization number who took one or more study drug doses and who had one or more post-baseline on-treatment primary efficacy assessments. The ADHD-RS-IV [16] was developed to measure behavior in children with ADHD. In this study, the ADHD-RS-AP, which consists of 18 items reflecting current ADHD symptoms based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria, was used. The adult prompts create a semi-structured measurement allowing clinicians to probe the extent, frequency, breadth, severity, and consequences of adult ADHD [17]. Items are scored on 4-point scales [0 (no symptoms) to 3 (severe symptoms)]; total score ranges from 0 to 54. The scale is divided into two subscales, hyperactivity/impulsivity and inattentiveness, consisting of nine items each. The ADHD-RS-AP was completed at each visit by an experienced clinician who was certified on the scale.
The key secondary efficacy endpoint was Clinical Global Impressions-Improvement (CGI-I) score at week 4. The CGI-I measures improvement in global symptom severity on a 7-point scale [1 (very much improved) to 7 (very much worse)] [18], with improvement measured against baseline CGI-Severity (CGI-S) scores. The CGI-I and CGI-S were completed by clinicians experienced in evaluating adult ADHD. Other secondary efficacy assessments included changes from baseline at week 4 on the subscales of ADHD-RS-AP and global functioning improvement based on the dichotomized CGI-I.
2.4 Safety Endpoints
Safety and tolerability assessments included treatment-emergent AEs (TEAEs), vital sign and weight changes, Columbia-Suicide Severity Rating Scale (C-SSRS) responses, and electrocardiogram results. All AEs were collected from the time of informed consent until the end of follow-up. Treatment-emergent AEs were defined as AEs occurring during double-blind treatment that (1) had start dates on or after the first study drug dose or (2) had start dates before the first study drug dose but that increased in severity on or after the date of the first study drug dose. Vital signs and weight were assessed at screening, baseline, and all visits through week 4/final on-treatment assessment, as were responses on the C-SSRS, which is a semi-structured interview that assesses suicidal ideation and behavior and non-suicidal self-injurious behavior [19]. Vital signs were assessed after approximately 3 min of rest with participants in a seated position and consisted of three assessments taken at approximately 2-min intervals. The 12-lead electrocardiogram was assessed at screening, baseline, week 2, and week 4/final on-treatment assessment after 3 min of rest; the baseline assessment consisted of three recordings taken at 3-min intervals.
2.5 Data Presentation and Statistics
Sample size was estimated for the primary efficacy comparison using nQuery Advisor® 7.0 (Statistical Solutions Ltd, Cork, Ireland). To detect a treatment difference of 7.0 [assumed common standard deviation (SD), 11.6] for the ADHD-RS-AP total score change from baseline, 60 participants per group were needed to provide 90% power for a two-sided t test (α = 0.05). Therefore, a total of 180 participants (60 for each SHP465 MAS dose and 60 for placebo) needed to be randomized. With an expected post-randomization dropout rate of 30%, the randomization target was set at 258 participants. The estimated treatment difference and SD corresponded to an effect size (ES) of 0.6 and was based on a previously completed phase III study of SHP465 MAS [15].
All statistical analyses of efficacy were conducted in the full analysis set. The primary efficacy endpoint (ADHD-RS-AP total score change from baseline at week 4) was analyzed using a linear mixed-effects model for repeated measures, with treatment group, visit, and the interaction of treatment group with the visit as factors; baseline ADHD-RS-AP total score as a covariate; and the interaction of baseline ADHD-RS-AP total score with the visit adjusted in the model. The null hypothesis was that there was no difference in the mean change from baseline at week 4 in the ADHD-RS-AP total score between each SHP465 MAS treatment group and the placebo group. The primary contrast was each dose of SHP465 MAS vs. placebo. To compare SHP465 MAS with placebo, the least-squares (LS) mean treatment difference for the change from baseline at week 4 and the corresponding 95% confidence interval and p value were calculated; ES (the LS mean difference divided by the square root of the variance estimate at week 4) was also calculated. Two sensitivity analysis models were used to examine the robustness of the primary analysis. These models assumed different missing not at random mechanisms and were within the pattern-mixture model framework.
The key secondary efficacy endpoint, CGI-I score at week 4, was analyzed using the same analysis methods described for the primary efficacy endpoint. Other assessments of efficacy were changes from baseline at week 4 on the ADHD-RS-AP subscales and dichotomized improvement on the CGI-I. Changes in ADHD-RS-AP subscales from baseline to week 4 were analyzed using the same mixed-effects model for repeated measures described for the primary efficacy endpoint. The dichotomized CGI-I analysis examined the percentage of participants who improved on the CGI-I at the final on-treatment assessment, with “improved” categorized as scores of 1 (very much improved) or 2 (much improved) and “not improved” categorized as scores of 3 (minimally improved) through 7 (very much worse). The percentage of improved participants for each SHP465 MAS dose vs. placebo was analyzed with a Cochran–Mantel–Haenszel test stratified by baseline CGI-S.
To preserve study-wide type I error at a two-sided α = 0.05 across the primary and key secondary efficacy assessments, a fixed-sequence test procedure was applied. In this procedure, each test was evaluated at a two-sided significance level of 0.05 and a later test could only be reported as significant if all earlier tests were significant. The order of assessment was as follows: 37.5 mg/day of SHP465 MAS vs. placebo for the ADHD-RS-AP total score change from baseline at week 4; 12.5 mg/day of SHP465 MAS vs. placebo for the ADHD-RS-AP total score change from baseline at week 4; 37.5 mg/day of SHP465 MAS vs. placebo for the CGI-I score at week 4; and 12.5 mg/day of SHP465 MAS vs. placebo for the CGI-I score at week 4. The study was not powered for the assessment of change from baseline at week 4 in ADHD-RS-AP subscale scores or for dichotomized improvement on the CGI-I at the final on-treatment assessment, and these endpoints were not included in the testing strategy. Therefore, all reported p values for these endpoints are nominal (unadjusted) and reported for descriptive purposes only. Safety and tolerability endpoints were assessed in the safety analysis set (all screened participants assigned a randomization number who took one or more study drug doses) and are reported using descriptive statistics.
3 Results
3.1 Participant Disposition and Demographics
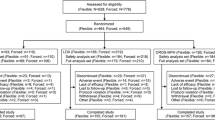
Participant disposition is summarized in Fig. 2. Of 369 screened participants, 275 were randomized (placebo, n = 91; 12.5 mg/day of SHP465 MAS, n = 92; 37.5 mg/day of SHP465 MAS, n = 92). A total of 271 and 263 participants, respectively, were included in the safety analysis set (placebo, n = 89; 12.5 mg/day of SHP465 MAS, n = 92; 37.5 mg/day of SHP465 MAS, n = 90) and full analysis set (placebo, n = 86; 12.5 mg/day of SHP465 MAS, n = 89; 37.5 mg/day of SHP465 MAS, n = 88).

Participant disposition. MAS mixed amphetamine salts
Demographic and clinical characteristics are summarized in Table 1. Participants were almost evenly divided across treatment arms. Overall, slightly more participants were men [150/271 (55.4%)] and a majority were white [81.5% (221/271)]. Mean age ranged from 32.4 years (37.5 mg/day of SHP465 MAS) to 34.5 years (placebo). Mean baseline ADHD-RS-AP total score ranged from 39.9 (37.5 mg/day of SHP465 MAS) to 40.4 (placebo); 80.8% (219/271) of participants were categorized as having the combined ADHD subtype.
3.2 Prior and Concomitant ADHD Medication Use
Prior ADHD medication was used in 71.9% (64/89), 62.0% (57/92), and 68.9% (62/90) of participants, respectively, in the placebo, 12.5-mg/day SHP465 MAS, and 37.5-mg/day SHP465 MAS treatment groups. The most frequently used ADHD medications (reported by ≥5% of participants in any treatment group) were immediate-release MAS [placebo, 44.9% (40/89); 12.5 mg/day of SHP465 MAS, 38.0% (35/92); 37.5 mg/day of SHP465 MAS, 32.2% (29/90)], methylphenidate [33.7% (30/89); 29.3% (27/92); 37.8% (34/90)], lisdexamfetamine [22.5% (20/89); 18.5% (17/92); 20.0% (18/90)], atomoxetine [5.6% (5/89); 9.8% (9/92); 10.0% (9/90)], and bupropion [2.2% (2/89); 4.3% (4/92); 5.6% (5/90)].
Concomitant medications were used by 64.0% (57/89), 52.2% (48/92), and 60.0% (54/90) of participants, respectively, in the placebo, 12.5-mg/day of SHP465 MAS, and 37.5-mg/day SHP465 MAS treatment groups. The most frequently reported concomitant medications (reported by ≥5% of participants in any treatment group) were ibuprofen [placebo, 15.7% (14/89); 12.5 mg/day of SHP465 MAS, 14.1% (13/92); 37.5 mg/day of SHP465 MAS, 15.6% (14/90)], multivitamins [9.0% (8/89); 6.5% (6/92); 11.1% (10/90)], acetaminophen [7.9% (7/89); 7.6% (7/92); 5.6% (5/90)], and loratadine [5.6% (5/89); 1.1% (1/92); 3.3% (3/90)]. Immediate-release MAS [placebo, 2.2% (2/89); 12.5 mg/day of SHP465 MAS, 3.3% (3/92); 37.5 mg/day of SHP465 MAS, 5.6% (5/90)] and lisdexamfetamine [placebo, 3.4% (3/89); 12.5 mg/day of SHP465 MAS, 1.1% (1/92); 37.5 mg/day of SHP465 MAS, 1.1% (1/90)] were the only concomitant ADHD medications used by ≥2% of participants in any treatment group. These instances were defined as protocol deviations or violations, and the numbers of participants taking these medications was low and comparable across treatment groups.
3.3 Efficacy
3.3.1 Primary Endpoint
Mean ADHD-RS-AP total scores decreased with all study treatments (Fig. 3a). Inferential analysis indicated that LS mean ± standard error of the mean changes from baseline in ADHD-RS-AP total score at week 4 were −18.5 ± 1.31 and −23.8 ± 1.34 in the 12.5- and 37.5-mg/day SHP465 MAS treatment groups, respectively; these reductions from baseline in ADHD-RS-AP total scores were statistically significantly greater than with placebo (−10.4 ± 1.33; both p < 0.001; Table 2). For the 12.5- and 37.5-mg/day SHP465 MAS treatment groups, respectively, ES was 0.67 and 1.11 (Table 2). Sensitivity analyses supported findings of the primary efficacy analysis (see Electronic Supplementary Material).

Attention-Deficit/Hyperactivity Disorder Rating Scale with Adult Prompts (ADHD-RS-AP) total score (a) and Clinical Global Impressions-Improvement (CGI-I) score (b) by treatment week. MAS mixed amphetamine salts, SD standard deviation; *p < 0.001 for the least-squares mean treatment difference vs. placebo for the change from baseline in the ADHD-RS-AP total score at week 4 (based on the mixed-effects model for repeated measures, including treatment, visit, and the interaction of treatment with the visit as factors; with the relevant baseline score as a covariate; and with an adjustment for the interaction of the baseline score with the visit); † p < 0.001 for the least-squares mean treatment difference vs. placebo for the CGI-I score at week 4 (based on the mixed-effects model for repeated measures, including treatment, visit, and the interaction of treatment with the visit as factors; with the relevant baseline score as a covariate; and with an adjustment for the interaction of the baseline score with the visit)
3.3.2 Secondary Endpoints
Mean CGI-I score decreased during the study in all treatment groups (Fig. 3b). Least-squares mean ± standard error of the mean CGI-I scores at week 4 (the key secondary endpoint) were 2.4 ± 0.12 and 1.9 ± 0.13 in the 12.5- and 37.5-mg/day SHP465 MAS treatment groups, respectively; these scores were statistically significantly lower than placebo (3.1 ± 0.12; both p < 0.001; Table 2). For 12.5- and 37.5-mg/day SHP465 MAS treatment groups, respectively, ES was 0.68 and 1.11 (Table 2).
Other secondary efficacy results are summarized in Table 2. The LS mean treatment difference for the change from baseline at week 4 on the ADHD-RS-AP hyperactivity/impulsivity subscale favored 12.5 and 37.5 mg/day of SHP465 MAS over placebo (both nominal p < 0.001; ES = 0.56 and 0.91, respectively; Table 2). Similarly, LS mean treatment differences for the change from baseline at week 4 on the ADHD-RS-AP inattentiveness subscale favored 12.5 and 37.5 mg/day of SHP465 MAS over placebo (both nominal p < 0.001; ES = 0.70 and 1.19, respectively; Table 2). At the final on-treatment assessment, the percentage of participants categorized as improved on CGI-I was higher with 12.5 mg/day of SHP465 MAS [55.1% (49/89)] and 37.5 mg/day of SHP465 MAS [75.0% (66/88)] than with placebo [30.2% (26/86); both nominal p < 0.001 based on the Cochran–Mantel–Haenszel test].
3.4 Safety and Tolerability
3.4.1 Adverse Events
The frequency of TEAEs was higher with SHP465 MAS than with placebo (Table 3). The most frequently reported TEAEs (those reported by >5% of participants in either SHP465 MAS treatment group) were decreased appetite, dry mouth, insomnia, headache, anxiety, initial insomnia, irritability, and bruxism. Most TEAEs were of mild-to-moderate intensity; no serious TEAEs were reported. Severe TEAEs were reported by two (2.2%) participants in the placebo treatment group, one (1.1%) participant in the 12.5-mg/day SHP465 MAS treatment group, and five (5.6%) participants in the 37.5-mg/day SHP465 MAS treatment group (see Table 3 footnote for a complete list). Treatment-emergent AEs leading to withdrawal of the study drug were reported by seven (7.6%) and five (5.6%) participants in the 12.5- and 37.50-mg/day SHP465 MAS treatment groups, respectively (see Table 3 footnote for a complete list).
3.4.2 Vital Signs
Mean ± SD changes in pulse, SBP, and DBP, respectively were numerically greater with both SHP465 MAS treatment groups (12.5 mg/day: 3.3 ± 10.52 bpm, 0.2 ± 7.24 and 1.0 ± 7.46 mmHg; 37.5 mg/day: 7.1 ± 11.48 bpm, 1.7 ± 9.99, and 2.8 ± 7.90 mmHg) compared with placebo (0.1 ± 8.35 bpm, −0.8 ± 9.99, and 0.1 ± 7.72 mmHg) at the final on-treatment assessment (Table 3). Increases in blood pressure and pulse with SHP465 MAS were greater with 37.5 mg/day of SHP465 MAS than with 12.5 mg/day of SHP465 MAS.
3.4.3 Other Safety and Tolerability Endpoints
At the final on-treatment assessment, mean ± SD weight and BMI increases were observed with placebo (0.30 ± 1.484 kg and 0.11 ± 0.510 kg/m2, respectively) and mean decreases were observed with SHP465 MAS (12.5 mg/day: −0.97 ± 1.523 kg and −0.33 ± 0.519 kg/m2; 37.5 mg/day: −1.65 ± 2.333 kg and −0.56 ± 0.777 kg/m2); changes in weight and BMI with 37.5 mg/day of SHP465 MAS were greater than those with 12.5 mg/day of SHP465 MAS (Table 3). At the final on-treatment assessment, mean ± SD changes in the Fridericia-corrected QT interval were −1.7 ± 11.80 ms with placebo, −3.0 ± 10.72 ms with 12.5 mg/day of SHP465 MAS, and −1.6 ± 13.70 ms with 37.5 mg/day of SHP465 MAS. No participant in any treatment group experienced a Fridericia-corrected QT ≥500 ms or had a change from baseline of ≥60 ms in Fridericia-corrected QT.
During the baseline assessment, one participant in the placebo group reported a lifetime history of a suicide attempt on the C-SSRS. However, no participant in any treatment group had a positive response for the on-study assessments of the C-SSRS.
4 Discussion
In this study, SHP465 MAS (12.5 and 37.5 mg/day) exhibited efficacy that was superior to placebo for reducing the ADHD-RS-AP total score (the primary efficacy endpoint), with a robust ES of 1.11 for 37.5 mg/day of SHP465 MAS. The effect of 12.5 mg/day of SHP465 MAS was numerically lower than 37.5 mg/day of SHP465 MAS. The efficacy of SHP465 MAS as measured by the primary efficacy assessment was supported by the results of the key secondary endpoint (CGI-I score reduction at week 4) and other efficacy endpoints (ADHD-RS-AP subscale score changes from baseline at week 4 and dichotomized CGI-I improvement at the final on-treatment assessment). These findings suggest that SHP465 MAS, at 12.5 and 37.5 mg/day, is an effective treatment in adults with ADHD.
The magnitude of the placebo-subtracted treatment effects observed in this study (12.5 mg/day of SHP465 MAS, −8.1; 37.5 mg/day of SHP465 MAS, −13.4) is within a range that is comparable to other studies of SHP465 MAS in adults with ADHD [13, 15]. In a 7-week dose-optimization (12.5–75 mg/day SHP465 MAS) study, the LS mean treatment difference for the ADHD-RS-IV total score reduction from baseline was −8.1 at the study endpoint [13]. In a 6-week forced-dose study, the LS mean treatment difference for ADHD-RS-IV total score reductions from baseline at the end of study ranged from −9.9 with 25 mg/day of SHP465 MAS (ES, 0.85) to −11.2 with 75 mg/day of SHP465 MAS (ES, 0.96) [15]. Although the effects of SHP465 MAS in this study are generally aligned with previous reports, the ES of 1.11 observed for 37.5 mg/day of SHP465 MAS exceeds that of previous reports in adults [13, 15]. Although it cannot be stated that dose-dependent treatment effects were observed in this study (the SHP465 MAS doses used in the study were not compared statistically), it is apparent that the placebo-subtracted treatment difference and ES for 37.5 mg/day of SHP465 MAS numerically exceeded those of 12.5 mg/day of SHP465 MAS. This difference could be clinically meaningful.
Although direct comparisons cannot be made between agents across different studies, the magnitude of effect observed in the current study is within a range observed for other long-acting psychostimulants in adults with ADHD [20, 21]. A 4-week fixed-dose study of lisdexamfetamine dimesylate in adults with ADHD reported an ES range of 0.73 for lisdexamfetamine dimesylate 30 mg/day to 0.99 for lisdexamfetamine dimesylate 70 mg/day. In a 4-week forced-dose study of MAS XR, the mean ES reported for MAS XR (20–60 mg/day) was 0.80. In further support of the robust ES observed in this study, a meta-analysis of psychostimulant treatment effects on the ADHD-RS total score from nine studies in adults with ADHD reported a mean ES of 0.89 [22].
The short-term safety and tolerability of SHP465 MAS in this study are consistent with observations from short-term placebo-controlled studies of SHP465 MAS [13, 15] and other long-acting psychostimulants in adults with ADHD [20, 21, 23, 24]. The most frequently reported TEAEs with SHP465 MAS (those occurring at a frequency of >5% and at twice the rate of placebo) in this study—decreased appetite, dry mouth, insomnia, headache, anxiety, initial insomnia, irritability, and bruxism—were consistent with those reported in other phase III studies of SHP465 MAS in adults with ADHD [13, 15]. These TEAEs have also been reported with other long-acting psychostimulants [20, 21, 23, 24]. In previously published short-term phase III studies, SHP465 MAS also increased pulse (range: 0.9–5.4 bpm), SBP (range −0.3 to 1.5 mmHg), and DBP (range −0.3 to 1.8 mmHg), and decreased weight (range −2.1 to –3.7 kg) [13, 15], which is consistent with reports of other long-acting psychostimulants [extended-release dexmethylphenidate (3.1–6.0 bpm; SBP −0.5 mmHg; DBP 1.0 mmHg); lisdexamfetamine (pulse 2.8–5.2 bpm; SBP 0.3–1.3 mmHg; DBP 0.8–1.6 mmHg); MAS XR (pulse 4.2–6.2 bpm; SBP 0.3–4.3 mmHg); osmotic controlled-release oral delivery system methylphenidate (pulse 3.6 bpm; SBP −1.2 mmHg; DBP 1.1 mmHg)] [20, 21, 23, 24].
The findings of this study should be considered in light of potential limitations. Although SHP465 MAS was titrated in the study, the forced-dose titration design used in this study is not consistent with the flexible-dose treatment strategies employed in clinical settings. As such, participants could have been maintained on doses that were suboptimal in terms of maximizing the balance between efficacy and safety/tolerability. Furthermore, a majority of study participants [67.5% (183/271)] had an ADHD medication treatment history, with the most frequently taken medications being stimulants. Because it is beyond the scope of this primary report to examine the role of treatment history on response to SHP465 MAS, it is not known how previous stimulant treatment would influence SHP465 MAS efficacy or tolerability.
Additionally, because the study was not powered for the assessment of changes in ADHD-RS-AP subscale scores or of dichotomized CGI-I improvement, the nominal p-values reported for those endpoints should be considered descriptive. It should also be noted that the 4-week duration of treatment was relatively short and does not allow for assessment of the long-term safety and tolerability of SHP465 MAS or of long-term clinical outcomes regarding ADHD symptoms. In addition, the current population was predominantly white and excluded individuals with comorbid psychiatric disorders. As such, generalizing these findings to a more diverse population of adults with ADHD should be considered cautiously because comorbidities are common in ADHD [25, 26] and their effects on the efficacy, tolerability, and safety of SHP465 MAS are not known. Last, the study also excluded individuals who met BMI criteria for being very severely obese (BMI ≥ 40 kg/m2). Although the mean BMI at baseline in this study indicates that on average study participants met BMI criteria for being overweight (BMI between 25 and 30 kg/m2), the exclusion of a proportion of obese individuals could limit the generalizability of these data because of the increased proportion of adults diagnosed with ADHD who meet criteria for obesity compared with adults who have not been diagnosed with ADHD [27].
5 Conclusions
The burden of illness of adult ADHD warrants efficacious treatment options that address ADHD symptoms. In this study, SHP465 MAS at a low dose of 12.5 mg/day was effective compared with placebo in alleviating adult ADHD symptoms. A dose of 37.5 mg/day of SHP465 MAS also demonstrated clinical efficacy, with an ES of 1.11. The safety profile of SHP465 MAS was consistent with previous observations in studies of adult ADHD treated with SHP465 MAS and other psychostimulants.
References
Chan E, Fogler JM, Hammerness PG. Treatment of attention-deficit/hyperactivity disorder in adolescents: a systematic review. JAMA. 2016;315(18):1997–2008.
Briars L, Todd T. A review of pharmacological management of attention-deficit/hyperactivity disorder. J Pediatr Pharmacol Ther. 2016;21(3):192–206.
Asherson P, Buitelaar J, Faraone SV, et al. Adult attention-deficit hyperactivity disorder: key conceptual issues. Lancet Psychiatry. 2016;3(6):568–78.
Bonvicini C, Faraone SV, Scassellati C. Attention-deficit hyperactivity disorder in adults: a systematic review and meta-analysis of genetic, pharmacogenetic and biochemical studies. Mol Psychiatry. 2016;21(7):872–84.
Rostain AL. Attention-deficit/hyperactivity disorder in adults: evidence-based recommendations for management. Postgrad Med. 2008;120(3):27–38.
Kooij SJ, Bejerot S, Blackwell A, et al. European consensus statement on diagnosis and treatment of adult ADHD: the European Network Adult ADHD. BMC Psychiatry. 2010;10:67.
Pliszka S, AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894–921.
Thomas M, Rostain A, Prevatt F. ADHD diagnosis and treatment in college students and young adults. Adolesc Med State Art Rev. 2013;24(3):659–79.
Jain R, Katic A. Current and investigational medication delivery systems for treating attention-deficit/hyperactivity disorder. Prim Care Companion CNS Disord. 2016;18(4). doi:10.4088/PCC.16r01979.
McGough JJ. Treatment controversies in adult ADHD. Am J Psychiatry. 2016;173(10):960–6.
Adler L. Adult ADHD. Washington, DC: Institute on Adult Presentation of Childhood Disorders, American Academy of Child and Adolescent Psychiatry; 2004.
Ermer JC, Shojaei A, Pennick M, et al. Bioavailability of triple-bead mixed amphetamine salts compared with a dose-augmentation strategy of mixed amphetamine salts extended release plus mixed amphetamine salts immediate release. Curr Med Res Opin. 2007;23(5):1067–75.
Spencer TJ, Adler LA, Weisler RH, et al. Triple-bead mixed amphetamine salts (SPD465), a novel, enhanced extended-release amphetamine formulation for the treatment of adults with ADHD: a randomized, double-blind, multicenter, placebo-controlled study. J Clin Psychiatry. 2008;69:1437–48.
Adler LA, Frick G, Yan B. A long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD. J Atten Disord. 2017. doi:10.1177/1087054717696770 (Epub ahead of print).
Frick G, Yan B, Adler LA. Triple-bead mixed amphetamine salts (SHP465) in adults with attention-deficit/hyperactivity disorder: results of a phase 3, double-blind, randomized, forced-dose trial. J Atten Disord. 2017. doi:10.1177/1087054717696771 (Epub ahead of print).
DuPaul GJ, Power TJ, Anastopoulos AD, et al. ADHD Rating Scale-IV: checklists, norms, and clinical interpretation. New York (NY): Guilford Press; 1998.
Goodman DW. ADHD in adults: update for clinicians on diagnosis and assessment. Prim Psychiatry. 2009;16(11):21–30.
Guy W. Clinical Global Impressions. Rockville: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse and Mental Health Administration; NIMH Psychopharmacology Research Branch; 1976.
Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–77.
Adler LA, Goodman DW, Kollins SH, et al. Double-blind, placebo-controlled study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69(9):1364–73.
Weisler RH, Biederman J, Spencer TJ, et al. Mixed amphetamine salts extended-release in the treatment of adult ADHD: a randomized, controlled trial. CNS Spectr. 2006;11(8):625–39.
Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71:754–63.
Adler LA, Zimmerman B, Starr HL, et al. Efficacy and safety of OROS methylphenidate in adults with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, double-blind, parallel group, dose-escalation study. J Clin Psychopharmacol. 2009;29(3):239–47.
Spencer TJ, Adler LA, McGough JJ, et al. Efficacy and safety of dexmethylphenidate extended-release capsules in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2007;61(12):1380–7.
Mao AR, Findling RL. Comorbidities in adult attention-deficit/hyperactivity disorder: a practical guide to diagnosis in primary care. Postgrad Med. 2014;126(5):42–51.
Kooij JJ, Huss M, Asherson P, et al. Distinguishing comorbidity and successful management of adult ADHD. J Atten Disord. 2012;16(5 Suppl.):3S–19S.
Cortese S, Moreira-Maia CR, St Fleur D, et al. Association between ADHD and obesity: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(1):34–43.
Acknowledgements
Under the direction of the authors, writing assistance was provided by Madhura Mehta, PhD, and Craig Slawecki, PhD (employees of Complete Healthcare Communications, LLC (Chadds Ford, PA, USA). Shire Development LLC (Lexington, MA, USA) provided funding to Complete Healthcare Communications for support in writing and editing this manuscript. Editorial assistance in the form of proofreading, copyediting, and fact checking was also provided by Complete Healthcare Communications. Shailesh Desai, PhD, from Shire, reviewed and edited the manuscript for scientific accuracy.
Author information
Authors and Affiliations
Contributions
RHW, MG, and VA were study investigators. They contributed to the development of the manuscript and reviewed and approved the final version of the manuscript. BY directed the statistical analysis of the data, contributed to the development of the manuscript, and reviewed and approved the final version of the manuscript. MY, MJ, and BR were involved in the design and execution of the study, contributed to the development of the manuscript, and reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Funding
The study was funded by Shire Development LLC (Lexington, MA, USA). Shire Development LLC provided funding to Complete Healthcare Communications, LLC (Chadds Ford, PA, USA) for support in writing and editing this manuscript and provided payment for open-access fees for this publication. The sponsor was involved in the study design, data collection and analysis, and data interpretation. The sponsor was also involved in the writing of the manuscript and in the decision to submit the article for publication, but the final content and decision to submit the manuscript to the CNS Drugs was made by the authors.
Conflict of interest
Richard H. Weisler in the last year has served as a consultant to Alcobra, AltheaDx Inc., Ironshore Pharmaceuticals, Lundbeck, Major League Baseball (certified clinician), National Football League (certified clinician), Neos Therapeutics, Nestlé Health Science-Pamlab Inc., Otsuka America Pharmaceuticals, Rhodes Pharmaceuticals, Shire, Sunovion, Supernus Pharmaceuticals, Takeda, and Validus; has received research support from Alcobra, Allergan, Daiichi Sankyo Pharma Genomind, Janssen, Merck, Neurim, Otsuka America Pharmaceuticals, Roche-Genentech, Shire, and Theravance; and has participated in speakers bureaus for Alcobra, Lundbeck, Neos Therapeutics, Otsuka America Pharmaceuticals, Rhodes Pharmaceuticals, Shire, Sunovion, and Validus. Michael Greenbaum has participated in a speaker’s bureau for Shire, and has received research support from Allergan, Lundbeck, Medgenics, Shire, Supernus, Sunovion, and Takeda. Valerie Arnold has received honoraria from Ironshore, Neos, Rho, and Shire; has served as a consultant for Ironshore; has participated in speaker’s bureaus for Takeda; and holds stock and/or stock options in Supernus. Brian Yan, Ming Yu, Margo Jaffee, and Brigitte Robertson are employees of Shire and hold stock and/or stock options in Shire.
Ethics approval
The study protocol, final approved informed consent document, and all supporting information were submitted to and approved by a central respective institutional review board (Copernicus Group Independent Review Board; Durham, NC, USA) and by the US Food and Drug Administration, as appropriate, before study initiation.
Consent to participate
All participants provided written informed consent before taking part in study procedures. The study was conducted in accordance with the International Conference on Harmonisation and Good Clinical Practice and the principles of the Declaration of Helsinki.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Weisler, R.H., Greenbaum, M., Arnold, V. et al. Efficacy and Safety of SHP465 Mixed Amphetamine Salts in the Treatment of Attention-Deficit/Hyperactivity Disorder in Adults: Results of a Randomized, Double-Blind, Placebo-Controlled, Forced-Dose Clinical Study. CNS Drugs 31, 685–697 (2017). https://doi.org/10.1007/s40263-017-0455-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0455-7