
Abstract
Citicoline is the generic name of the pharmaceutical substance that chemically is cytidine-5′-diphosphocholine (CDP-choline), which is identical to the natural intracellular precursor of phospholipid phosphatidylcholine. Following injection or ingestion, citicoline is believed to undergo quick hydrolysis and dephosphorylation to yield cytidine and choline, which then enter the brain separately and are used to resynthesize CDP-choline inside brain cells. Neuroprotective activity of citicoline has been repeatedly shown in preclinical models of brain ischaemia and trauma, but two recent, large, pivotal clinical trials have revealed no benefits in ischaemic stroke and traumatic brain injury. However, the substance seems to be beneficial in some slowly advancing neurodegenerative disorders such as glaucoma and mild vascular cognitive impairment. This paper critically discusses issues related to the clinical pharmacology of citicoline, including its pharmacokinetics/biotransformation and pharmacodynamics/mode of action. It is concluded that at present, there is no adequate description of the mechanism(s) of the pharmacological actions of this substance. The possibility should be considered and tested that, in spite of apparently fast catabolism, the intact citicoline molecule or the phosphorylated intermediate products of its hydrolysis, cytidine monophosphate and phosphocholine, are pharmacologically active.
Similar content being viewed by others


Citicoline is chemically identical to CDP-choline, the natural precursor of the major cell membrane phospholipid phosphatidylcholine. |
Given orally or by injection, citicoline is non-toxic and very well tolerated. |
Preclinical experiments with various models of central neurodegenerative diseases have shown that citicoline displays significant neuroprotective properties. |
However, recent large and well-controlled data have shown no benefit from citicoline in acute ischaemic stroke and traumatic brain injury. |
The pharmacological actions of citicoline in the central nervous system seem to be pleiotropic and involve, amongst other things, modulation of some kinases and sirtuin-1. However, our understanding of the mechanisms involved is, at most, fragmentary. |
1 Introduction
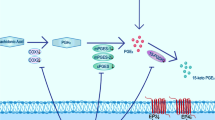
Citicoline is the generic name, or the International Nonproprietary Name (INN) of cytidine-5′-diphosphocholine (CDP-choline, CDPCho), a pharmaceutical substance that is chemically identical to the naturally occurring metabolite, which plays crucial role in the synthesis of phospholipids. The work of Kennedy and collaborators in the 1950s showed that this phosphorylated choline nucleotide is a precursor of glycerophospholipid phosphatidylcholine (PC) [1]. PC and its twin compound phosphatidylethanolamine (PEt) are the two most abundant phospholipids in eukaryotic cells, accounting for more than half of the total phospholipid content in membranes. The pathway of de novo synthesis of PC, the CDP-choline pathway (Fig. 1), includes the enzymes cytidine kinase (CK), choline phosphate cytidilyltransferase (CCT) and CDP-choline:1,2-diacylglycerol choline phosphotransferase (CPT) [2]. These enzymes are located intracellularly, in the endoplasmic reticulum and cell nucleus [3]. Choline that is used for synthesis of phosphatidylcholine through the CDP-choline pathway is derived from transport of exogenous choline into the cell or from phospholipase D-mediated turnover of PC [4].

The cytidine-5′-diphosphocholine (CDP-choline) pathway of enzymatic synthesis of phosphatidylcholine. ADP adenosine diphosphate, ATP adenosine triphosphate, CCT choline phosphate cytidilyltransferase, CK cytidine kinase, CMP cytidine monophosphate, CPT CDP-choline:1,2-diacylglycerol choline phosphotransferase, CTP cytidine triphosphate, DAG 1,2-dicacylglycerol, PC phosphatidylcholine, PP i pyrophosphate
The search for relevant literature—non-clinical as well as clinical—was performed in the PubMed, Scopus and Web of Science databases, using the term ‘CDP-choline’, its variants (‘CDPcholine’, ‘cytidine-diphosphocholine’, etc.) and ‘citicoline’. Whereas ‘CDP-choline’ designates the natural metabolite synthesized inside the cells (a substance of endogenous origin), the term ‘citicoline’ did not exist until the substance became used as the drug, which occurred in the 1970s. Notwithstanding, some papers currently indexed under the term ‘citicoline’ are dated earlier; some of them (e.g. the paper by Berger and Gimenez [5], describing crystallization of CDP-choline from yeast) could be qualified as concerning citicoline (the compound synthesized exogenously), but others (e.g. the paper by Ansell and Bayliss [6], reporting on the concentration of endogenous CDP-choline in the rat brain) undoubtedly concern CDP-choline (the compound synthesized endogenously). The difference between CDP-choline (synthesized endogenously) and citicoline (synthesized exogenously) is not trivial, as will be discussed later.
In the Anglophone medical literature, the use citicoline as a drug was advocated as early as 1974, by Manaka et al. [7] from Japan, for the treatment of Parkinson’s disease. A few years later, the idea of using it as a neuroprotectant stemmed from the observation, made by Horrocks and collaborators [8], of the reversibility of phosphotransferase enzymes in the brain. The first patent for the use of CDP-ethanolamine, alone or in combination with CDP-choline, to reverse neurodegenerative diseases was issued in 1981 [9]. Since that time, hundreds of studies concerning various aspects of the preclinical and clinical pharmacology of citicoline have been published. Several relevant reviews have also appeared in high-ranking journals, including at least five during the last 4 years [10–14]. However, the subject of citicoline’s mode of action is far from being clarified. The present review is an attempt to classify the major issues concerning the neuroprotective properties of citicoline into three categories: facts, i.e. those that are solved with an acceptable degree of certainty; doubts, i.e. those that are likely misinterpreted; and unresolved issues, i.e. those that remain unexplained. Needless to say, such classification is subjective: facts, doubts and unresolved issues are, in many cases, intermingled.
2 Facts
Citicoline displays negligible toxicity. The compound is quickly catabolized (Fig. 2), and the products arising are subsequently available for diverse biosynthetic pathways and ultimately excreted as carbon dioxide. The lack of acute and chronic toxicity of citicoline has been repeatedly confirmed in rodents and dogs (see the most recent report by Schauss et al. [15] and the references quoted therein). An impressive example is the median lethal dose (LD50) of an acute single intravenous application of citicoline, which equals 4,600 and 4,150 mg/kg in mice and rats, respectively. The LD50 for ingested citicoline is even higher at approximately 8 g/kg in both mice and rats. For comparison, in mice, the LD50 of an acute single intravenous dose of sodium chloride is 645 mg/kg, and that of vitamin C is 518 mg/kg. In a 90-day rat oral toxicity study of 100–1,000 mg/kg daily doses, increases in serum creatinine and in renal tubular mineralization, likely caused by phosphate liberation from citicoline, were found, without concomitant degenerative or inflammatory reactions.

Presumed catabolism of citicoline (Cyt-P-P-Cho) in the rodent intravascular compartment. In the first step, hydrolysis of the pyrophosphate bridge takes place. In the second step, cytidine monophosphate (Cyt-P) and phosphocholine (Cho-P) are dephosphorylated to cytidine (Cyt) and choline (Cho), respectively; supposedly, a large part of the liberated choline is taken up by the liver (which may explain the unexpectedly low cholinergic toxicity of citicoline)
The usual daily therapeutic dosage of citicoline in humans is 500–2,000 mg—that is, 7–28 mg/kg in a person of average bodyweight (70 kg). Data from clinical trials have corroborated preclinical toxicological findings, revealing a favourable safety profile, with only a few reports of adverse events, mostly related to digestive disturbances following oral intake. In adult and elderly stroke patients, the drug has lacked significant adverse events (see, for example, the study by Cho and Kim [16] in 4,191 Korean stroke patients), and a meta-analysis of placebo-controlled trials has shown that the overall frequency of adverse effects was comparable between groups comprising 1,652 actively treated and 686 placebo-treated subjects [17]. There are no data concerning the effects of liver or kidney insufficiency on the safety profile and pharmacokinetics of citicoline although, on the basis of the aforementioned toxicology data, an enhanced threat of hyperphosphataemia may be predicted in patients with kidney failure.
Upon administration, citicoline is relatively quickly catabolized and is the source of choline that appears in the blood. Administered parenterally or orally, citicoline is relatively quickly (i.e. within minutes rather than hours) converted to its cholinergic and pyrimidinergic catabolites. In the perfused rat liver, citicoline disappeared from the perfusate within 10 min [18, 19]. Since phosphorylated substrates are considered unable to penetrate cell membranes, it is usually assumed that cytidine monophosphate (CMP) and phosphocholine (PCho) yielded from hydrolysis of citicoline are further dephosphorylated by phosphatases in blood plasma. In agreement with this assumption, citicoline given orally to rats produced pronounced increases in plasma cytidine and choline, although it should be noted that the rise of cytidine was several-fold larger than that of choline [20]. A recently published paper [21] reported on the use of a liquid chromatography electrospray ionization tandem mass spectrometry (LC–ESI–MS/MS) method to evaluate the pharmacokinetics of choline in blood from human volunteers following ingestion of 1,000 mg citicoline tablets. Biphasic concentration–time curves of choline in plasma have been recorded, with a large peak of 2 μg/mL at approximately 2 h and a second, slightly smaller but much broader peak with a maximum at approximately 24 h following ingestion. Unfortunately, no data on the initial plasma level of choline were shown, which makes the whole picture a bit unclear. One older source reported an arithmetic mean plasma choline level oscillating around 1.36 μg/mL [22] in a healthy human subject, and this was confirmed recently [23].
Is citicoline carcinogenic? In a wide variety of cancers, choline phospholipid metabolism is altered in such a way that cancer cells display elevated levels of phosphocholine, as well as total choline-containing compounds [24]. One may therefore pose a question as to whether increasing choline exposure doesn’t induce carcinogenesis and/or accelerate cancer growth. However, choline chloride has displayed no mutagenic potential when tested in vitro (using Ames testing, yeast gene conversion, clastogenicity and sister chromatid exchange) [25], and similar negative data have been obtained for citicoline [26]. Moreover, epidemiological data have shown that the associations between choline intake and cancer—if any—are weak. For example, Johansson et al. [27] found that elevated plasma concentrations of choline may be associated with a slightly increased risk of prostate cancer, but a similar (even slightly stronger) association has been found for vitamin B2. On the other hand, Lee et al. [28] did not find any association between choline (or betaine) intake and the risk of colorectal cancer, whereas Xu et al. [29] found that dietary choline intake was inversely associated with breast cancer risk.
Citicoline is neuroprotective in various animal (preclinical) experimental paradigms. The compound has offered marked neuroprotection in several in vitro and in vivo models of acute and chronic brain ischaemic and neurodegenerative diseases, including brain hypoxia, ischaemia and intracerebral haemorrhage (reviewed by Adibhatla and Hatcher [30]), brain and spinal cord trauma [31], in vitro glutamate excitotoxicity [32, 33] and in vivo amyloid toxicity [34]. However, the mechanisms of this neuroprotection are far from being understood.
One major effect of citicoline is believed to be stimulation of the synthesis and increase in the content of brain phospholipids. Increases in brain phospholipids following oral administration of citicoline have also been observed in humans, with use of phosphorous magnetic resonance spectroscopy [35]. The relevant hypothesis assumes that the citicoline breakdown products cytidine and choline enter the brain separately and, inside brain cells, they act as substrates for resynthesis of CDP-choline. This, in turn, is believed to result in slowing down of phospholipid breakdown and acceleration of phospholipid resynthesis necessary for membrane repair [36]. However, since citicoline is devoid of cholinergic toxicity (see below), a significant rise in brain choline following therapeutic doses of citicoline in humans does not seem probable—indeed, a decrease in choline in the brains of older subjects and no change in those of younger subjects have been observed following oral citicoline, with use of proton magnetic resonance spectroscopy (MRS) [37].
The other mechanisms suggested to be involved in the neuroprotective effects of citicoline in stroke models include prevention of activation of phospholipase A2 (PLA2) [38]. The related effects comprise attenuation of the increase in hydroxyl radical generation, preventing loss of cardiolipin (an exclusive inner mitochondrial membrane phospholipid essential for mitochondrial electron transport, which is degraded in response to cellular insults and disrupts the mitochondrial respiratory chain). In aged rats, an increase in the brain level of platelet-activating factor (a bioactive phospholipid implicated in neuronal excitotoxic death) has also been noted [39]. In rats, attenuation of mitogen-activated protein kinases (MAPKs) and caspase activation have been observed following citicoline administration [40, 41]. Last, but not least, according to the most recent report [42], treatment with citicoline has been found to increase sirtuin-1 (SIRT1) protein levels in cultured neurons, in circulating blood mononuclear cells and in the brain. This effect seems to be of critical importance for neuroprotection in experimental stroke because sirtinol, a specific inhibitor of SIRT1 which, by itself, does not influence infarct volume, has been shown to abolish the neuroprotection offered by citicoline. Citicoline displayed a potent synergistic effect with resveratrol (which is known to be a SIRT1 activator), leading to a 60 % reduction in the experimental infarct volume in rats when both drugs were used in doses that were individually ineffective. Moreover, citicoline was ineffective in SIRT1 knock-out homozygotic mice subjected to focal brain ischaemia. However, detailed mechanistic explanations for all of these effects are lacking. For example, there is no explanation as to how citicoline administration leads to attenuation of MAPK activity and increases sirtuin-1 protein content in brain tissues; in particular, does the drug act extracellularly, or is resynthesis of CDP-choline inside brain cells a prerequisite?
Citicoline is not beneficial in patients with stroke and traumatic brain injury. Positive results of preclinical studies with animal models of neurodegenerative diseases have prompted clinical trials with citicoline as a treatment for human brain diseases. Whereas several previous small clinical studies had achieved promising results, two recent large randomized multicenter trials—the COBRIT (Citicoline Brain Injury Treatment) trial performed in 1,213 patients with traumatic brain injury [43], and the international, randomized, multicentre, placebo-controlled sequential ICTUS (International Citicoline Trial on Acute Stroke) trial performed in 2,298 patients with moderate-to-severe acute ischaemic stroke [44]—led to the conclusion that citicoline is not efficacious in these clinical settings. The negative outcomes of these studies were deemed surprising and prompted a few comments, which focused mostly, although not exclusively, on methodological aspects of the evaluation of the clinical effects of the drug [45–47]. What was not commented on was the lack of a mechanistic explanation for the putative neuroprotective properties of citicoline.
Citicoline treatment seems beneficial in some chronic neurodegenerative diseases. Some recent data are suggestive that prolonged intake of citicoline, given orally or by injection, may be significantly effective in certain slowly developing neurodegenerative diseases. One is glaucoma, currently considered a neurodegenerative disease, which involves the entire central visual pathway. In glaucoma patients with moderate visual defects, citicoline treatment improved retinal function and neural conduction, and continuation of treatment for 2–8 years significantly slowed, stabilized or even improved glaucomatous visual dysfunction [48, 49]. The other is mild vascular cognitive impairment. In the open IDEALE (Studio di Intervento nel Decadimento Vascolare Lieve) study [50], oral citicoline taken for up to 9 months significantly improved the Mini-Mental State Examination score and positively influenced mood; the latter effect could have been related to increases in noradrenaline and dopamine levels, which would be expected on the basis of animal experiments (see the paper by Rejdak et al. [51] and the references cited therein).
Also, it has recently been shown that in sub-acute ischaemic cerebrovascular disease, administration of citicoline in an intravenous dose of 2,000 mg for 5 or 10 days improves functional independence and reduces the burden of care [52]. The uniqueness of citicoline may lie not only in its negligible toxicity and virtual lack of side effects but also in the fact that it appears to deliver a significant subjective improvement and mood-enhancing effect.
3 Doubts
Where is citicoline catabolized? While it seems reasonable to assume that, at least in rodents, citicoline given systemically is quickly hydrolysed and further dephosphorylated, the particulars of this decomposition process are uncertain. Does it take place in blood plasma or in other location(s)? Of note is that, following injection or ingestion of citicoline in the rat, the increase in plasma cytidine was, on a molar basis, several times larger than the concomitant increase in plasma choline [20]. What happened to the ‘surplus’ choline? Was it taken up by the liver? Does the several-fold ‘domination’ of the cytidine increase over the choline increase provide an explanation for the lack of cholinergic toxicity of citicoline versus the toxic effects of equimolar choline (see below)?
How is citicoline catabolized? In humans, an oral challenge with citicoline was accompanied by an increase in plasma uridine instead of cytidine [53]. In this last study, the participants took oral citicoline doses of up to 4 g (still much less per kilogram of bodyweight than the doses used in most rodent experiments), and significant dose-related increases in blood plasma uridine were observed. The data resembled the rat data reported previously by the same laboratory [20] in that the magnitude of the choline increases was several times smaller than the magnitude of the pyrimidine increase. Choline increments obtained after 2 or 4 g doses were comparable, on a molar basis, to those seen after similar doses of choline chloride, although the peaks were delayed by one or more hours. The authors failed, however, to detect any significant quantities of cytidine in human blood, either before or after citicoline intake. They interpreted this finding as evidence that the human gastrointestinal tract and liver quantitatively transform cytidine liberated from citicoline to circulating uridine. Would this indicate that hydrolysis of citicoline to CMP and PCho and consecutive dephosphorylations of these products to cytidine and choline occur almost immediately, followed by conversion of cytidine to uridine? But why is the final effect—namely, an increase in circulating choline—delayed by one or more hours?
Resynthesis of CDP-choline in the brain following citicoline intake and the pharmacodynamics of citicoline. It is usually assumed that following citicoline intake, cytidine and choline enter brain cells separately and are used for intracellular synthesis of CDP-choline [36]. For example, in a recent paper, Ramos-Cabrer et al. [54] stated “It is believed that when citicoline is exogenously administered as sodium salt, it is hydrolysed into choline and cytidine to be re-synthesized later in the brain…”. These statements seem to imply that resynthesis of citicoline from cytidine and choline in the brain is the only event—or at least the most important event—of systemic citicoline application.
At this point, the difference between citicoline (synthesized exogenously) and CDP-choline (synthesized endogenously) acquires its key importance, for at least three reasons. First, it must be appreciated that only a minor fraction of the choline dose administered as citicoline enters the brain. Tolvanen et al. [55] investigated the biodistribution and biokinetics of [11C]choline (a radio-pharmaceutical used for oncological positron emission tomography [PET] studies) in rats and humans following intravenous injection. They found that the highest uptake of the tracer was by the kidney, lung and adrenal glands, whereas the brain cortex and cerebellum were the organs taking up less than 0.1 % of the tracer dose. Second, stimulation following citicoline intake of at least two other major synthetic pathways of choline in the brain, one leading to betaine (an important donor of methyl groups) and the other leading to acetylcholine (an important CNS neurotransmitter), should also be taken into account [56]. Third, the ischaemic heart is the source of increased choline in the blood of patients with an acute coronary syndrome related to coronary plaque instability [57]. Moreover, the authors mention small unpublished pilot studies in which elevated levels of whole blood choline were also found in patients with stroke or cerebral ischaemia in combination with advanced plaques in the carotid artery. It is reasonable to assume that degradation of membrane phospholipids and elevation of blood choline levels occur in all cases of brain ischaemia, i.e. ischaemic stroke. Is there any difference between the metabolic effects of choline increases in plasma consequential to citicoline intake and those resulting from a heart attack or stroke?
Why were preclinical results with citicoline for stroke not reproduced in the clinical setting? The systematic review and meta-analysis of data obtained with preclinical models of embolic stoke [58] provided evidence that citicoline does indeed deliver some neuroprotection; however, the effect is stronger for infarct volume reduction and more limited for neurological outcome. The authors concluded that factors shown to be important for translation into human studies are multiple-dose administration, large infarct size and/or neurological deficit. However, the most important cause of the irreproducibility of preclinical results in a clinical setting may be the use of excessively large doses of citicoline in most animal experiments. For example, in a series of papers authored by Savci and collaborators (see the paper by Eyigor et al. [59] and the references quoted therein), various cardiovascular and endocrine effects of large, although subtoxic, doses of citicoline (0.5–2 g/kg) given intravenously were described. Among them were an increase in blood pressure and large rises in plasma levels of catecholamines and several pituitary hormones, including vasopressin and oxytocin. Evidence of histaminergic system involvement in the responses to citicoline has also been presented [60]. Can these effects be of potential benefit, e.g. in hypovolaemic shock (as suggested by Savci et al. [61]), or are they instead an early sign of toxicity? (On the other hand, it would be of importance to find an explanation for the observation that in rats—both normotensive and haemorrhagic—these massive doses of citicoline increased blood pressure, whereas no such effect occurred following equivalent doses of either cytidine or choline.)
Similar doubts may be raised against the recent report by Gutiérrez-Fernández et al. [62], who compared the effects of citicoline (1,000 mg/kg intraperitoneally) and recombinant tissue plasminogen activator [rt-PA] (5 mg/kg intravenously) in a rat model of embolic stroke. The problem with this paper was that whereas the rt-PA dose used per kg of bodyweight was 5 to 8 times the dose used in the treatment of human embolic stroke (0.6–1.1 mg/kg intravenously; see Wardlaw et al. [63]), the citicoline dose was 15 to 30 times the dose used in clinical trials of citicoline in stroke. The rationale for investigating the effects of injecting citicoline in doses corresponding to 30–150 g per person (assuming an average human bodyweight of 70 kg) is doubtful.
4 Unresolved Issues
Why is citicoline so much less toxic than choline? The acute toxicity of citicoline and choline after oral and intravenous application was compared by Agut et al. [64]. These authors concluded that CDP-choline given either orally or intravenously did not cause any cholinergic intoxication in the treated groups, whereas such toxic effects were observed after administration of an equimolar dose of choline. Apparently, CDP-choline given by the oral or intravenous route yields toxicological consequences that are different from those yielded by choline. A mechanistic interpretation of the substantially lower toxicity of citicoline compared with that of choline is lacking to date.
Can intact citicoline be delivered orally? Yashima et al. [65] used citicoline doubly-labelled with carbon-14 at the methyl groups of choline and tritium at C5 of pyrimidine, and obtained evidence of CDP-choline being broken down in the intestine, resulting in release of choline and cytidine, likely due to the actions of intestinal esterases and pyrophosphatases. However, the authors concluded that “these split compounds as well as the original CDP-choline may be absorbed from the intestinal mucosa as such”. Apparently, they could not rule out the possibility that following oral intake, some fraction of intact citicoline is absorbed.
Is citicoline a prodrug or an active compound? The consequences of the assumed fast hydrolysis and subsequent dephosphorylation of citicoline after injection or oral intake are usually interpreted in terms of a prodrug, which is administered in an inactive or less than fully active form and is subsequently metabolically converted (bioactivated) to the active pharmacological agents, cytidine and choline. However, one may assume that the reverse is true, i.e. the most active form is unhydrolysed (intact) citicoline, whereas cytidine and choline are its pharmacologically less active metabolites. Although the prodrug concept clearly prevails in the scientific literature, several observations may indicate that intact citicoline molecules, or perhaps CMP and/or PCho intermediates, display their own activities, which are different from and/or more potent than the actions of cytidine and choline.
One reason for not discarding the idea of intact citicoline being significantly neuroprotective is the magnitude of the protective effect of citicoline in vitro versus in vivo. Neuroprotective effects in vitro occurred upon exposure of retinal cells or brain neurons to citicoline concentrations as low as submicromolar to micromolar [66, 67], whereas in the in vivo (animal) experiments, the minimal doses necessary to produce appreciable neuroprotection were within the range of 0.3–1 g/kg [68], an amount that could be equivalent to 0.6–2 mmol/kg, assuming even distribution throughout the body. Thus, citicoline seems to be a neuroprotectant that acts weakly in vivo but is much more potent in vitro.
The other reason concerns the activity of liposomal citicoline. A few studies have indicated that in experimental ischaemic stroke, liposomal formulations of citicoline are significantly more neuroprotective (i.e. by means of decreasing the ischaemic infarct volume) than equivalent doses of free citicoline (see, for example Fresta and Puglisi [69, 70] and Adibhatla et al. [69, 70]). The most recent study of this kind [54] indicated that liposomal citicoline is more neuroprotective than the equivalent intravenous dose of the free drug which, in turn, is more neuroprotective than the equivalent intraperitoneal dose. Although other explanations may be speculated upon, the aforementioned result is compatible with the idea that intact citicoline is pharmacologically more active than its metabolites.
Does intact citicoline modulate some kinases? Interesting observations have been reported on the modulation by citicoline of the activity/expression of some protein kinases involved in neuronal death—namely, MAPKs—in the postischaemic brain [40] and extracellular signal-regulated kinase 1/2 (ERK1/2) in the rat retina after kainic acid (KA) treatment [71]. Modulation of activity of cellular kinases and, in particular, members of the ERK/MAPK family, occurs through transduction of extracellular signals [72, 73]. Perhaps the neuroprotective actions of citicoline are exerted not by its hydrolysis products but by the unhydrolysed molecules acting extracellularly as signalling molecules. In this context, one should consider the possibility of binding of citicoline to plasma proteins such as albumin. Albumin binding has been reported for cytidine [74] and also for sphingosylphosphorylcholine [75], a compound remotely similar to citicoline. If intact citicoline binds to albumin, its hydrolysis could be retarded and its action as a signalling molecule could be prolonged.
5 Conclusions
In spite of the negative results of recent pivotal studies in acute ischaemic stroke and traumatic brain injury, there is continuing interest in the neuroprotective properties of citicoline. The drug is non-toxic, and numerous preclinical data support the view that it displays neuroprotective properties. However, no adequate mechanistic explanation for these observations has ever been provided.
The most frequently presented explanation for the neuroprotective effects of citicoline on the brain is based on the assumption that it is a prodrug which, following injection or ingestion, is sequentially hydrolysed and dephosphorylated, finally, to cytidine (or uridine in humans) and choline. Then these two metabolites separately enter the brain tissues and are used to resynthesize CDP-choline, which exerts neuroprotection intracellularly by supporting biosynthesis of cellular phospholipids. An alternative explanation—i.e. that unhydrolysed citicoline, or perhaps phosphocholine and/or cytidine monophosphate, are pharmacologically active metabolites of citicoline—has never been considered.
References
Weiss SB, Smith SW, Kennedy EP. The enzymatic formation of lecithin from cytidine diphosphate choline and d-1,2-diglyceride. J Biol Chem. 1958;231:53–64.
Gibellini F, Smith TK. The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010;62:414–28.
Hunt AN, Clark GT, Attard GS, Postle AD. Highly saturated endonuclear phosphatidylcholine is synthesized in situ and colocated with CDP-choline pathway enzymes. J Biol Chem. 2001;276:8492–9.
Kent C, Carman GM. Interactions among pathways for phosphatidylcholine metabolism, CTP synthesis and secretion through the Golgi apparatus. Trends Biochem Sci. 1999;24:146–50.
Berger L, Gimenez WT. Crystallization of cytidine diphosphate choline from yeast. Science. 1956;124:81.
Ansell GB, Bayliss BJ. The cytidine diphosphate choline content of rat brain. Biochem J. 1961;78:209–13.
Manaka S, Sano K, Fuchinoue T, Sekino H. Mechanism of action CDP-choline in parkinsonism. Experientia. 1974;30:179–80.
Horrocks LA, Dorman RV, Dabrowiecki Z, Goracci G, Porcellati G. CDPcholine and CDPethanolamine prevent the release of free fatty acids during brain ischemia. Prog Lipid Res. 1981;20:531–4.
Horrocks LA, Dorman RV, Dabrowiecki ZM. Therapeutic agents for preventing phospholipid degradation and free fatty acid proliferation. United States Patent. 1981 no: 4,386,078.
García-Cobos R, Frank-García A, Gutiérrez-Fernández M, Díez-Tejedor E. Citicoline, use in cognitive decline: vascular and degenerative. J Neurol Sci. 2010;299:188–92.
Alvarez-Sabín J, Román GC. Citicoline in vascular cognitive impairment and vascular dementia after stroke. Stroke. 2011;42:S40–3.
Arenth PM, Russell KC, Ricker JH, Zafonte RD. CDP-choline as a biological supplement during neurorecovery: a focused review. PMR. 2011;6:S123–31.
Hurtado O, Lizasoain I, Moro MÁ. Neuroprotection and recovery: recent data at the bench on citicoline. Stroke. 2011;42:S33–5.
Secades JJ. Probably role of citicoline in stroke rehabilitation: review of the literature. Rev Neurol. 2012;54:173–9.
Schauss AG, Somfai-Relle S, Financsek I, Glavits R, Parent SC, Endres JR, Varga T, Szücs Z, Clewell A. Single- and repeated-dose oral toxicity studies of citicoline free-base (choline cytidine 5′-pyrophosphate) in Sprague-Dawley rats. Int J Toxicol. 2009;28:479–87.
Cho HJ, Kim YJ. Efficacy and safety of oral citicoline in acute ischemic stroke: drug surveillance study in 4,191 cases. Methods Find Exp Clin Pharmacol. 2009;31:171–6.
Dávalos A, Castillo J, Alvarez-Sabín J, Secades JJ, Mercadal J, López S, Cobo E, Warach S, Sherman D, Clark WM, Lozano R. Oral citicoline in acute ischemic stroke: an individual patient data pooling analysis of clinical trials. Stroke. 2002;33:2850–7.
Galletti P, De Rosa M, Cotticelli MG, Morana A, Vaccaro R, Zappia V. Biochemical rationale for the use of CDPcholine in traumatic brain injury: pharmacokinetics of the orally administered drug. J Neurol Sci. 1991;103:S19–25.
Galletti P, De Rosa M, Nappi MA, Pontoni G, del Piano L, Salluzzo A, Zappia V. Transport and metabolism of double-labelled CDPcholine in mammalian tissues. Biochem Pharmacol. 1985;34:4121–30.
López-Coviella I, Agut J, Savci V, Ortiz JA, Wurtman RJ. Evidence that 5′-cytidinediphospho-choline can affect brain phospholipid composition by increasing choline and cytidine plasma levels. J Neurochem. 1995;65:889–94.
Sarkar AK, Ghosh D, Haldar D, Sarkar P, Gupta B, Dastidar SG, Pal TK. A rapid LC-ESI-MS/MS method for the quantitation of choline, an active metabolite of citicoline: application to in vivo pharmacokinetic and bioequivalence study in Indian healthy male volunteers. J Pharm Biomed Anal. 2012;71:144–7.
Bligh J. The level of free choline in plasma. J Physiol. 1951;117:234–40.
Adamczyk M, Brashear RJ, Mattingly PG. Choline concentration in normal blood donor and cardiac troponin-positive plasma samples. Clin Chem. 2006;52:2123–4.
Glunde K, Serkova NJ. Therapeutic targets and biomarkers identified in cancer choline phospholipid metabolism. Pharmacogenomics. 2006;7:1109–23.
EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Scientific opinion on safety and efficacy of choline chloride as a feed additive for all animal species. EFSA J. 2011;9:2353.
Food Safety Authority of Ireland. Safety assessment of citicoline. 2012. http://www.fsai.ie/uploadedFiles/Science_and_Health/Novel_Foods/Applications/2012%20Citicholine.pdf. Accessed 24 Oct 2012.
Johansson M, Van Guelpen B, Vollset SE, Hultdin J, Bergh A, Key T, Midttun O, Hallmans G, Ueland PM, Stattin P. One-carbon metabolism and prostate cancer risk: prospective investigation of seven circulating B vitamins and metabolites. Cancer Epidemiol Biomark Prev. 2009;18:1538–43.
Lee JE, Giovannucci E, Fuchs CS, Willett WC, Zeisel SH, Cho E. Choline and betaine intake and the risk of colorectal cancer in men. Cancer Epidemiol Biomark Prev. 2010;19:884–7.
Xu X, Gammon MD, Zeisel SH, Lee YL, Wetmur JG, Teitelbaum SL, Bradshaw PT, Neugut AI, Santella RM, Chen J. Choline metabolism and risk of breast cancer in a population-based study. FASEB J. 2008;22:2045–52.
Adibhatla RM, Hatcher JF. Cytidine 5′-diphosphocholine (CDP-choline) in stroke and other CNS disorders. Neurochem Res. 2005;30:15–23.
Yücel N, Cayli SR, Ateş O, Karadağ N, Firat S, Turköz Y. Evaluation of the neuroprotective effects of citicoline after experimental spinal cord injury: improved behavioral and neuroanatomical recovery. Neurochem Res. 2006;31:767–75.
Matyja E, Taraszewska A, Nagańska E, Grieb P, Rafałowska J. CDP-choline protects motor neurons against apoptotic changes in a model of chronic glutamate excitotoxicity in vitro. Folia Neuropathol. 2008;46:139–48.
Mir C, Clotet J, Aledo R, Durany N, Argemí J, Lozano R, Cervós-Navarro J, Casals N. CDP-choline prevents glutamate-mediated cell death in cerebellar granule neurons. J Mol Neurosci. 2003;20:53–60.
Alvarez XA, Sampedro C, Lozano R, Cacabelos R. Citicoline protects hippocampal neurons against apoptosis induced by brain beta-amyloid deposits plus cerebral hypoperfusion in rats. Methods Find Exp Clin Pharmacol. 1999;21:535–40.
Babb SM, Wald LL, Cohen BM, Villafuerte RA, Gruber SA, Yurgelun-Todd DA, Renshaw PF. Chronic citicoline increases phosphodiesters in the brains of healthy older subjects: an in vivo phosphorus magnetic resonance spectroscopy study. Psychopharmacology (Berl). 2002;161:248–54.
D’Orlando KJ, Sandage BW Jr. Citicoline (CDP-choline): mechanisms of action and effects in ischemic brain injury. Neurol Res. 1995;17:281–4.
Babb SM, Appelmans KE, Renshaw PF, Wurtman RJ, Cohen BM. Differential effect of CDP-choline on brain cytosolic choline levels in younger and older subjects as measured by proton magnetic resonance spectroscopy. Psychopharmacology (Berl). 1996;127:88–94.
Adibhatla RM, Hatcher JF. Citicoline decreases phospholipase A2 stimulation and hydroxyl radical generation in transient cerebral ischemia. J Neurosci Res. 2003;73:308–15.
Giménez R, Aguilar J. Effects of CDP-choline administration on brain striatum platelet-activating factor in aging rats. Eur J Pharmacol. 1998;344:149–52.
Krupinski J, Slevin M, Badimon L. Citicoline inhibits MAP kinase signalling pathways after focal cerebral ischaemia. Neurochem Res. 2005;30:1067–73.
Krupinski J, Abudawood M, Matou-Nasri S, Al-Baradie R, Petcu E, Justicia C, Planas A, Liu D, Rovira N, Grau-Slevin M, Secades J, Slevin M. Citicoline induces angiogenesis improving survival of vascular/human brain microvessel endothelial cells through pathways involving ERK1/2 and insulin receptor substrate-1. Vasc Cell. 2012;4:20.
Hurtado O, Hernández-Jiménez M, Zarruk JG, Cuartero MI, Ballesteros I, Camarero G, Moraga A, Pradillo JM, Moro MA, Lizasoain I. Citicoline (CDP-choline) increases Sirtuin1 expression concomitant to neuroprotection in experimental stroke. J Neurochem. 2013;126:819–26.
Zafonte RD, Bagiella E, Ansel BM, Novack TA, Friedewald WT, Hesdorffer DC, Timmons SD, Jallo J, Eisenberg H, Hart T, Ricker JH, Diaz-Arrastia R, Merchant RE, Temkin NR, Melton S, Dikmen SS. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308:1993–2000.
Dávalos A, Alvarez-Sabín J, Castillo J, Díez-Tejedor E, Ferro J, Martínez-Vila E, Serena J, Segura T, Cruz VT, Masjuan J, Cobo E, Secades JJ, International Citicoline Trial on Acute Stroke (ICTUS) Trial Investigators. Citicoline in the treatment of acute ischaemic stroke: an international, randomised, multicentre, placebo-controlled study (ICTUS trial). Lancet. 2012;380:349–57.
Clark WM, Clark TD. Stroke: treatment for acute stroke—the end of the citicoline saga. Nat Rev Neurol. 2012;8:484–5.
Hankey GJ. How effective is citicoline for acute ischaemic stroke? Lancet. 2012;380:318–20.
Ruff RL, Riechers RG. Effective treatment of traumatic brain injury: learning from experience. JAMA. 2012;308:2032–3.
Parisi V, Coppola G, Centofanti M, Oddone F, Angrisani AM, Ziccardi L, Ricci B, Quaranta L, Manni G. Evidence of the neuroprotective role of citicoline in glaucoma patients. Prog Brain Res. 2008;173:541–54.
Ottobelli L, Manni GL, Centofanti M, Iester M, Allevena F, Rossetti L. Citicoline oral solution in glaucoma: is there a role in slowing disease progression? Ophthalmologica. 2013;229:219–26.
Putignano S, Gareri P, Castagna A, Cerqua G, Cervera P, Cotroneo AM, Fiorillo F, Grella R, Lacava R, Maddonni A, Marino S, Pluderi A, Putignano D, Rocca F. Retrospective and observational study to assess the efficacy of citicoline in elderly patients suffering from stupor related to complex geriatric syndrome. Clin Interv Aging. 2012;7:113–8.
Rejdak R, Toczołowski J, Solski J, Duma D, Grieb P. Citicoline treatment increases retinal dopamine content in rabbits. Ophthalmic Res. 2002;34:146–9.
Cotroneo AM, Castagna A, Putignano S, Lacava R, Fantò F, Monteleone F, Rocca F, Malara A, Gareri P. Effectiveness and safety of citicoline in mild vascular cognitive impairment: the IDEALE study. Clin Interv Aging. 2013;8:131–7.
Wurtman RJ, Regan M, Ulus I, Yu L. Effect of oral CDP-choline on plasma choline and uridine levels in humans. Biochem Pharmacol. 2000;60:989–92.
Ramos-Cabrer P, Agulla J, Argibay B, Pérez-Mato M, Castillo J. Serial MRI study of the enhanced therapeutic effects of liposome-encapsulated citicoline in cerebral ischemia. Int J Pharm. 2011;405:228–33.
Tolvanen T, Yli-Kerttula T, Ujula T, Autio A, Lehikoinen P, Minn H, Roivainen A. Biodistribution and radiation dosimetry of [(11)C]choline: a comparison between rat and human data. Eur J Nucl Med Mol Imaging. 2010;37:874–83.
Weiss GB. Metabolism and actions of CDP-choline as an endogenous compound and administered exogenously as citicoline. Life Sci. 1995;56:637–60.
Danne O, Möckel M. Choline in acute coronary syndrome: an emerging biomarker with implications for the integrated assessment of plaque vulnerability. Expert Rev Mol Diagn. 2010;10:159–71.
Bustamante A, Giralt D, Garcia-Bonilla L, Campos M, Rosell A, Montaner J. Citicoline in pre-clinical animal models of stroke: a meta-analysis shows the optimal neuroprotective profile and the missing steps for jumping into a stroke clinical trial. J Neurochem. 2012;123:217–25.
Eyigor O, Coskun C, Cavun S, Savci V. Intravenous CDP-choline activates neurons in supraoptic and paraventricular nuclei and induces hormone secretion. Brain Res Bull. 2012;87:286–94.
Jochem J, Savci V, Filiz N, Rybus-Kalinowska B, Fogel WA, Yalcin M. Involvement of the histaminergic system in cytidine 5′-diphosphocholine-induced reversal of critical haemorrhagic hypotension in rats. J Physiol Pharmacol. 2010;61:37–43.
Savci V, Goktalay G, Cansev M, Cavun S, Yilmaz MS, Ulus IH. Intravenously injected CDP-choline increases blood pressure and reverses hypotension in haemorrhagic shock: effect is mediated by central cholinergic activation. Eur J Pharmacol. 2003;468:129–39.
Gutiérrez-Fernández M, Leciñana MA, Rodríguez-Frutos B, Ramos-Cejudo J, Roda JM, Díez-Tejedor E. CDP-choline at high doses is as effective as i.v. thrombolysis in experimental animal stroke. Neurol Res. 2012;34:649–56.
Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, Cohen G. Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta-analysis. Lancet. 2012;379:2364–72.
Agut J, Font E, Sacristán A, Ortiz JA. Dissimilar effects in acute toxicity studies of CDP-choline and choline. Arzneimittelforschung. 1983;33:1016–8.
Yashima K, Takamatsu M, Okuda K. Intestinal absorption of cytidine diphosphate choline and its changes in the digestive tract. J Nutr Sci Vitaminol (Tokyo). 1975;21:49–60.
Mykita S, Golly F, Dreyfus H, Freysz L, Massarelli R. Effect of CDP-choline on hypocapnic neurons in culture. J Neurochem. 1986;47:223–31.
Oshitari T, Fujimoto N, Adachi-Usami E. Citicoline has a protective effect on damaged retinal ganglion cells in mouse culture retina. Neuroreport. 2002;13:2109–11.
Secades JJ, Frontera G. CDP-choline: pharmacological and clinical review. Methods Find Exp Clin Pharmacol. 1995;17B:1–54.
Fresta M, Puglisi G. Survival rate improvement in a rat ischemia model by long circulating liposomes containing cytidine-5I-diphosphate choline. Life Sci. 1997;61:1227–35.
Adibhatla RM, Hatcher JF, Tureyen K. CDP-choline liposomes provide significant reduction in infarction over free CDP-choline in stroke. Brain Res. 2005;1058:193–7.
Park CH, Kim YS, Cheon EW, Noh HS, Cho CH, Chung IY, Yoo JM, Kang SS, Choi WS, Cho GJ. Action of citicoline on rat retinal expression of extracellular-signal-regulated kinase (ERK1/2). Brain Res. 2006;1081:203–10.
Kyriakis JM. Making the connection: coupling of stress-activated ERK/MAPK (extracellular-signal-regulated kinase/mitogen-activated protein kinase) core signalling modules to extracellular stimuli and biological responses. Biochem Soc Symp. 1999;64:29–48.
Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–75.
Cui F, Wang J, Yao X, Wang L, Zhang Q, Qu G. Spectroscopic and molecular modeling studies of the interaction between cytidine and human serum albumin and its analytical application. Biopolymers. 2007;87:174–82.
Han M, Kim YL, Sacket SJ, Kim K, Kim HL, Jo JY, Ha NC, Im DS. Effect of direct albumin binding to sphingosylphosphorylcholine in Jurkat T cells. Prostaglandins Other Lipid Mediat. 2007;84:174–83.
Acknowledgements
Dr Grieb has no conflicts of interest that are directly relevant to the content of this review. No sources of funding were used in the preparation of the review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Grieb, P. Neuroprotective Properties of Citicoline: Facts, Doubts and Unresolved Issues. CNS Drugs 28, 185–193 (2014). https://doi.org/10.1007/s40263-014-0144-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-014-0144-8