
Abstract
Background
Emicizumab (ACE910) is a bispecific antibody mimicking the cofactor function of activated coagulation factor VIII. In phase I–I/II studies, emicizumab reduced the bleeding frequency in patients with severe hemophilia A, regardless of the presence of factor VIII inhibitors, at once-weekly subcutaneous doses of 0.3, 1, and 3 mg/kg.
Methods
Using the phase I–I/II study data, population pharmacokinetic and repeated time-to-event (RTTE) modeling were performed to quantitatively characterize the relationship between the pharmacokinetics of emicizumab and reduction in bleeding frequency. Simulations were then performed to identify the minimal exposure expected to achieve zero bleeding events for 1 year in at least 50% of patients and to select the dosing regimens to be tested in phase III studies.
Results
The RTTE model adequately predicted the bleeding onset over time as a function of plasma emicizumab concentration. Simulations suggested that plasma emicizumab concentrations of ≥ 45 μg/mL should result in zero bleeding events for 1 year in at least 50% of patients. This efficacious exposure provided the basis for selecting previously untested dosing regimens of 1.5 mg/kg once weekly, 3 mg/kg every 2 weeks, and 6 mg/kg every 4 weeks for phase III studies.
Conclusions
A pharmacometric approach guided the phase III dose selection of emicizumab in hemophilia A, without conducting a conventional dose-finding study. Phase III studies with the selected dosing regimens are currently ongoing. This case study indicates that a pharmacometric approach can substitute for a conventional dose-finding study in rare diseases and will streamline the drug development process.
Similar content being viewed by others


A repeated time-to-event model described the exposure-dependent, bleeding-prophylactic effect of emicizumab in patients with severe hemophilia A with or without factor VIII inhibitors. |
Model-based simulations enabled the selection of previously untested dosing regimens of emicizumab for phase III studies, without conducting a conventional dose-finding study. |
A pharmacometric analysis leveraging early-phase clinical study data can provide a substitute for a conventional dose-finding study in the development of new drugs in rare diseases. |
1 Introduction
Hemophilia A is an X-linked inherited bleeding disorder that occurs in approximately 1 in 5000 male births [1]. The disease is caused by a deficiency of coagulation factor VIII (FVIII). Approximately half of patients are classified as having a severe phenotype, defined as having < 1 IU/dL (< 1% of normal) of endogenous FVIII activity, which leads to higher bleeding frequency than moderate (1–5 IU/dL) or mild (> 5 to < 40 IU/dL) phenotypes [2,3,4]. The standard of care for hemophilia A includes episodic and prophylactic therapies to control bleeding with recombinant or plasma-derived FVIII. However, the prophylactic regimen, targeting a trough FVIII activity of ≥ 1 IU/dL, requires intravenous infusion of FVIII twice or more times per week due to its short elimination half-life (8–19 h) [4,5,6,7], which can impose a substantial burden of treatment on patients [2, 8, 9]. Moreover, anti-FVIII neutralizing alloantibodies (‘FVIII inhibitors’) may develop in up to approximately 30% of patients with severe hemophilia A receiving FVIII [10, 11], which renders treatment with FVIII ineffective. Bypassing agents, such as activated prothrombin complex concentrates and recombinant activated factor VII, are used for patients with FVIII inhibitors where immune tolerance induction against FVIII is not successful. However, their efficacy for the prevention and control of bleeding is suboptimal, and frequent intravenous infusions are required.
Emicizumab (ACE910) is a recombinant, humanized, bispecific monoclonal antibody that simultaneously binds to activated factor IX (FIXa) and factor X (FX), thereby mimicking the cofactor function of activated FVIII [12,13,14]. Non-clinical investigations have suggested that emicizumab can be administered subcutaneously, has a longer elimination half-life than existing treatments, is effective regardless of the presence or absence of FVIII inhibitors, and is not expected to induce FVIII inhibitors [12, 13, 15, 16]. Altogether, these characteristics could address an unmet need in hemophilia A treatment. In a single-ascending-dose phase I study in Japanese and Caucasian healthy volunteers, emicizumab demonstrated linear pharmacokinetics, an elimination half-life of approximately 4–5 weeks, pharmacokinetic similarity between Japanese and Caucasian populations, and a favorable safety profile at single subcutaneous (SC) doses of 0.001–1 mg/kg [17]. Subsequently, in a 12-week, multiple-ascending-dose phase I study and its long-term extension phase I/II study in Japanese patients with severe hemophilia A with or without FVIII inhibitors, emicizumab demonstrated linear pharmacokinetics, a favorable safety profile, and reduction in the individual patients’ annualized bleeding rates (ABRs), by 22.8–100% compared with their own historical data, at once-weekly (QW) SC doses of 0.3–3 mg/kg [18, 19].
This remarkable preliminary efficacy prompted the sponsors to seek innovative ways to shorten the overall development timeline, particularly for patients with FVIII inhibitors whose unmet medical need is higher. Demand for rapid development together with the limited number of patients with FVIII inhibitors precluded the conduct of an adequately powered, randomized, controlled dose-finding study (‘conventional dose-finding study’) before embarking on the phase III program. However, determining the dose–response relationship to support the selection of the dosing regimens to be tested in phase III studies, simply based on the observed data in the preceding phase I–I/II studies, was difficult due to the limited sample size and imbalanced baseline disease characteristics across the dosing groups [18].
A pharmacometric analysis presented herein was therefore performed to refine the relationship between the pharmacokinetics of emicizumab and reduction in bleeding frequency, and to guide the selection of phase III dosing regimens. In line with the preceding studies, a QW dosing regimen was initially explored for phase III dose selection. The possibility of reducing the dosing frequency to once every 2 weeks (Q2W) or every 4 weeks (Q4W), while maintaining similar efficacy and equivalent cumulative dose, was then investigated.
2 Methods
2.1 Clinical Studies, Subjects, and Data
The analysis dataset included 24 Japanese and 18 Caucasian healthy male adult volunteers who received a single SC administration of emicizumab at 0.01, 0.1, 0.3, or 1 mg/kg [17], and 18 Japanese male adult or adolescent patients with severe hemophilia A with or without FVIII inhibitors who received QW SC administrations of emicizumab at 0.3, 1, or 3 mg/kg (Cohorts 1, 2, and 3, respectively) [18, 19] (Table 1). Healthy volunteers who received 0.001 mg/kg of emicizumab were not included in the dataset because their plasma emicizumab concentrations were all below the lower limit of quantification (< 0.05 μg/mL). In the patient study, the initial doses for Cohorts 1 and 2 were single loading doses of 1 and 3 mg/kg, respectively. Before the start of emicizumab treatment, Cohort 1 had a higher median ABR and Cohort 3 had a higher proportion of patients with prophylaxis with coagulation factor treatment, resulting in imbalanced baseline disease characteristics across the three cohorts.
In these clinical studies, the plasma emicizumab concentration, activated partial thromboplastin time (aPTT), activated factor XI-triggered thrombin generation (TG), and anti-emicizumab antibodies were measured using previously described methods [17] at defined timepoints (see the Supplementary Methods in the Electronic Supplementary Material [ESM]). Bleeding event data were collected in patients prospectively during the study and retrospectively from their medical records for the 6 months prior to study enrollment. When a patient started experiencing any bleeding or bleeding-related symptoms (e.g., pain, swelling), regardless of the types (traumatic or non-traumatic), sites, and duration, it was reported as the onset of a bleeding event. Bleeding events that required episodic use of coagulation factor treatment were considered for the analysis.
Complete data from healthy volunteers and interim data from patients up to November 2014 were used for the analysis for phase III dose selection, and are presented here unless otherwise stated. Because the patient study is still ongoing, subsequently available data up to February 2016 [19] were used for validating the developed models (see ESM Methods).
2.2 Graphical Exposure-Response Exploration
Only patients were included in this analysis. The relationship between plasma emicizumab concentrations, aPTT, or TG and bleeding events during emicizumab treatment were graphically explored. Because these pharmacokinetic and pharmacodynamic measurements were collected at defined timepoints, they might not coincide with the onset of bleeding. Therefore, predictions of these markers at the onset times of each bleeding event were derived by linear interpolation and plotted on the observed individual time courses to investigate the dependency of bleeding onset on the pharmacokinetics or pharmacodynamics of emicizumab.
2.3 Population Pharmacokinetic (PopPK) Modeling
Both healthy volunteers and patients were included in this analysis. A one-compartment model with first-order absorption and elimination was employed for the structural model for SC dosing. Inter-individual variability following a log-normal distribution was assumed for each structural model parameter of clearance (CL/F), volume of distribution (V d/F), and first-order absorption half-life (t ½,abs). A combined additive-plus-proportional error model was assumed for the residual unexplained variability.
Covariate effects were modeled as follows:
For a continuous covariate:
For a categorical covariate:
where P i,t is the value of a structural model parameter at time t for subject i; P TV is the typical value of a structural model parameter; Cov i,t is the value of a continuous covariate or flag (0 or 1) for a categorical covariate at time t for subject i; CovTV is the typical value of a continuous covariate; θ Cov is the effect of a covariate on a structural model parameter (called allometric exponent if for body weight [BW]); and η i is the assigned inter-individual variability of a structural model parameter for subject i.
BW and anti-emicizumab neutralizing antibody (NAb) positivity were structural covariates requiring no statistical testing for inclusion in the model. The effects of BW on CL/F and V d/F were fixed to the allometric exponents of 0.75 and 1, respectively [20,21,22]. For one Caucasian volunteer who developed NAbs, the effect of NAb positivity on CL/F and its onset time following a single SC administration of emicizumab were estimated through model fitting using the MTIME/MPAST functionality of NONMEM® version 7.2.0 (ICON Development Solutions, Ellicott City, MD, USA) [23]. The significances of the effects of subject type (healthy volunteer or patient) and age on each structural model parameter were statistically investigated by likelihood ratio test with a forward inclusion criterion of P < 0.05 and a backward exclusion criterion of P ≥ 0.01. If the effect of subject type on a parameter was statistically significant, additional investigation on the effect of FVIII inhibitor status on the parameter was hierarchically made.
First-order conditional estimation with interaction method was used for maximum likelihood estimation. Model evaluation was made by goodness-of-fit plots, prediction-corrected visual predictive checks (pcVPCs) [24], and parametric bootstrapping [25].
2.4 Repeated Time-to-Event (RTTE) Modeling
RTTE modeling [26, 27] was used to characterize the exposure–response relationship of emicizumab in patients. Likelihood of having a bleeding event at a time was modeled with a time-varying hazard function consisting of a constant ‘baseline’ bleeding hazard (the bleeding hazard where no prophylaxis was given [λ]), assuming that bleeding events occurred independently from each other, the effects of emicizumab and another prophylactic treatment (given during the prior 6 months and/or a post-emicizumab follow-up after stopping emicizumab treatment) in an inhibitory E max (maximum effect) model, and a pharmacodynamic drug–drug interaction between emicizumab and another prophylactic treatment. Inter-individual variability following a log-normal distribution was assumed for λ and the plasma emicizumab concentration to reduce λ by half (EC50).
The RTTE model equations employed were as follows:
If a bleeding event is right-censored:
If a bleeding event occurs:
where h i,t is the bleeding hazard at time t for patient i; λ i is the λ for patient i; C emi,i,t is the PopPK model-derived plasma emicizumab concentration at time t for patient i; EC50,i is the EC50 for patient i; PLX i,t is the status of ongoing/no use of another prophylactic treatment at time t for patient i; θ PLX is the effect of another prophylactic treatment on bleeding hazard; t last,i,t is the onset time of latest bleeding event before time t (or the starting time of observation if no bleeding event had occurred) for patient i; H i,t is the cumulative bleeding hazard until time t since t last,i,t for patient i; L 0,i,t is the likelihood (survival function) of not having had a bleeding event until time t since t last,i,t for patient i; and L 1,i,t is the likelihood (probability density function) of having a bleeding event at time t since tlast,i,t for patient i (see ESM Appendix S1).
In the E max model, C emi was corrected by EC50 to enable combination of the effects of emicizumab and another prophylactic treatment as additive interaction [28,29,30]. The assumption of this type of interaction between emicizumab and another prophylactic treatment was based on the facts that one patient without FVIII inhibitors received prophylaxis with FVIII, the mechanism of action of which is similar to emicizumab, in the presence of emicizumab remaining in plasma during a post-emicizumab follow-up, and that no patient received prophylaxis with a bypassing agent in the presence of emicizumab (see ESM Methods).
FVIII inhibitor status, use of another prophylactic treatment, and types of the used prophylactic treatment may affect bleeding hazard. However, due to the strong correlation among these factors in the dataset, estimating their individual effects was difficult. Therefore, only the status of ongoing/no use of another prophylactic treatment, regardless of the drug type, was selected for a structural covariate to descriptively correct the difference in bleeding hazard between patients or periods with and without prophylaxis with coagulation factor treatment. The effects of FVIII inhibitor status and age on EC50 were explored by the same statistical approaches as those for the PopPK modeling.
First-order conditional estimation with Laplacian method was used for maximum likelihood estimation. Model evaluation was made by visual predictive checks (VPCs) and posterior predictive checks [31] on the cumulative numbers of bleeding events having occurred (‘cumulative bleeding counts’) until the last times of observation before and after the start of emicizumab treatment, and non-parametric bootstrapping to obtain the 95% confidence intervals (CIs) of each parameter estimate. Minimum and maximum were selected for the variability statistics plotted in the VPCs because the small number of patients in the multiple-ascending-dose study and its extension study, which decreased over time, did not allow proper derivation of generally used statistics (e.g., 5th and 95th percentiles) at later timepoints.
2.5 Simulation-Based Exploration of Target Efficacious Exposure and Doses
To illustrate the exposure-dependent reduction in bleeding frequency with emicizumab, individual cumulative bleeding counts for 52 weeks (‘annual bleeding rates’, equated with ABRs) and the proportions of patients with zero ABR were simulated at a series of constant plasma emicizumab concentrations (with no peak–trough fluctuations) using the developed RTTE model (see ESM Methods). The target efficacious exposure for guiding phase III dose selection was defined as the minimal concentration expected to achieve zero ABR in ≥ 50% of patients, which is comparable efficacy with prophylaxis with FVIII demonstrated in patients without FVIII inhibitors [32,33,34,35,36,37]. Thereafter, the plasma emicizumab concentration–time profiles and ABR distributions at potential dosing regimens to achieve the target efficacious exposure were simulated using the developed PopPK and RTTE models (see ESM Methods). Loading doses were selected to achieve the target efficacious exposure as rapidly as possible without exceeding the highest dose tested in the preceding studies.
2.6 Software
All the modeling and simulation analyses were performed using NONMEM® version 7.2.0 assisted by Perl-speaks-NONMEM version 3.5.3 (http://psn.sourceforge.net/).
3 Results
3.1 Graphical Exposure-Response Exploration
The graphical investigation indicated a plasma emicizumab concentration-dependent reduction in bleeding frequency (Fig. 1). In contrast, there were no clear correlations between either aPTT or TG and bleeding onset (ESM Fig. S1), possibly because these pharmacodynamic markers already achieved their maximal effects (plateaus) at low plasma emicizumab concentrations [18]. Therefore, plasma emicizumab concentration was selected as the predictor of bleeding onset for the subsequent pharmacometric analyses for phase III dose selection.

Individual time courses of plasma emicizumab concentration (solid lines) with bleeding onset (open circles) during emicizumab treatment. Data during a post-emicizumab follow-up are not plotted
3.2 PopPK Modeling
A PopPK model was developed to describe the pharmacokinetics of emicizumab following SC dosing (Table 2). Patients had 1.26-fold the CL/F and 1.19-fold the V d/F of healthy volunteers. The model-derived elimination half-lives for healthy volunteers and patients were 31.8 and 30.1 days, respectively. FVIII inhibitor status and age (over the subjects’ age range of ≥ 12 years) did not significantly affect either structural model parameter. This PopPK model adequately described the observed plasma emicizumab concentration–time profiles (Fig. 2, ESM Figs. S2, S3, S4).

Prediction-corrected visual predictive check plots for the developed population pharmacokinetic model. Data for the first 16 weeks (a) and the entire time of observation (b) after receiving a single (for healthy volunteers) or the first (for patients) subcutaneous administration of emicizumab are presented. Red solid line indicates the observed median, red dashed lines indicate the observed 5th (lower) and 95th (upper) percentiles, and shaded areas indicate the simulated 95% confidence intervals of the 5th percentile (blue, bottom), median (red, middle), and 95th percentile (blue, top)
3.3 RTTE Modeling
A RTTE model was developed to characterize the exposure–response relationship of emicizumab (Table 3). The bleeding hazard in patients or during periods with prophylaxis with coagulation factor treatment was estimated to be 23.9% lower, with a wide 95% CI (0.313–66.2% lower), than those without prophylaxis. No significant effects of FVIII inhibitor status or age on EC50 were found. In conjunction with the PopPK findings, application of the same dose of emicizumab to either adult or adolescent patient populations with or without FVIII inhibitors was supported. This RTTE model adequately predicted the bleeding onset over time both before and after the start of emicizumab treatment (Fig. 3, ESM Figs. S5 and S6).

Visual predictive check plots for the developed repeated time-to-event model. Data before (a) and after (b) the start of emicizumab treatment are presented. Red solid line indicates the observed median, red dashed lines indicate the observed minimum (lower) and maximum (upper), and shaded areas indicate the simulated 95% confidence intervals of the minimum (blue, bottom), median (red, middle), and maximum (blue, top)
3.4 Identification of Target Efficacious Exposure
The simulated median ABR was 22 in the absence of emicizumab or any other prophylactic treatment (Fig. 4). An increased plasma emicizumab concentration was associated with decreased median ABR and the increased proportion of patients with zero ABR. A plasma emicizumab concentration of 45 μg/mL was identified as the minimal concentration to achieve zero ABR in ≥ 50% of patients and selected for the target efficacious exposure to be achieved in phase III studies.

Simulated relationship of plasma emicizumab concentration with annual bleeding rate (a) or the proportion of patients with an annual bleeding rate of zero (b). Plotted plasma emicizumab concentrations include every 2 μg/mL from 0 to 20 μg/mL, every 5 μg/mL from 20 to 100 μg/mL, and every 10 μg/mL from 100 to 200 μg/mL. a Open circles and solid line indicate the simulated median, and shaded area indicates the simulated 5th to 95th percentile range; b open circles and solid line indicate the simulated proportion, and dashed line indicates the target proportion of 50%
3.5 Identification of Target Efficacious Doses
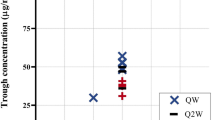
Based on the PopPK simulations, a maintenance dose of 1.5 mg/kg was identified as the minimal dose to provide a median steady-state trough concentration (C trough,ss) of ≥ 45 μg/mL with QW dosing (Fig. 5a). Median trough concentrations were predicted to reach 45 μg/mL following repeated loading doses of 3 mg/kg QW for the first 4 weeks. A maintenance dose of 3 mg/kg Q2W was also predicted to achieve ≥ 45 μg/mL as median C trough,ss (Fig. 5b). With a lower dosing frequency associated with a larger peak–trough fluctuation, a maintenance dose of 6 mg/kg Q4W was predicted to result in a median C trough,ss slightly lower than 45 μg/mL (Fig. 5c). However, the differences in the plasma emicizumab concentration–time profiles among the QW, Q2W, and Q4W dosing regimens were not expected to impact the ABR distribution based on the simulations using the integrated PopPK-RTTE model (Fig. 6), possibly because higher peak concentrations (accompanying lower trough concentrations) counterbalance the overall bleeding-prophylactic effect. Of note, for all the QW, Q2W, and Q4W dosing regimens, even the peak concentrations were predicted to remain below 120 μg/mL (the observed mean C trough,ss at 3 mg/kg QW which was the highest dosing regimen tested previously and deemed to be well-tolerated [19]) at steady state, and the peak–trough fluctuations were limited (ESM Table S1). These results supported the selection of the maintenance doses of 1.5 mg/kg QW, 3 mg/kg Q2W, and 6 mg/kg Q4W following the common 4-week loading dose of 3 mg/kg QW for phase III dosing regimens.

Simulated time courses of plasma emicizumab concentration at the selected phase III dosing regimens of a repeated subcutaneous loading dose of 3 mg/kg once weekly for first 4 weeks followed by subcutaneous maintenance doses of 1.5 mg/kg once weekly (a), 3 mg/kg every 2 weeks (b), and 6 mg/kg every 4 weeks (c). Solid line indicates the simulated median profile, open circles indicate the simulated median trough concentrations, shaded area indicates the simulated 5th to 95th percentile range, and dashed line indicates the target efficacious exposure of 45 μg/mL

Simulated distributions of annual bleeding rate at the selected phase III dosing regimens of a repeated subcutaneous loading dose of 3 mg/kg once weekly for first 4 weeks followed by subcutaneous maintenance doses of 1.5 mg/kg once weekly (a), 3 mg/kg every 2 weeks (b), and 6 mg/kg every 4 weeks (c). Data for annual bleeding rates of more than 8 are not plotted
4 Discussion
The dose–exposure–response relationship of emicizumab in patients with severe hemophilia A with or without FVIII inhibitors was quantitatively characterized by a pharmacometric analysis. This analysis identified the target efficacious exposure and formed the primary rationale for selecting the phase III dosing regimens, which had not been tested in the preceding phase I–I/II studies, without conducting a conventional dose-finding study.
Disease phenotypes for patients with severe hemophilia A receiving prophylaxis with FVIII vary over time in association with the time course of replaced FVIII activity. A negative binomial (NB) model demonstrated that a ‘summarized’ exposure variable of time spent with FVIII activity of < 1 IU/dL is associated with increased bleeding frequency [38]. Meanwhile, clinically relevant exposure will not be limited to the trough side [39]. In the present research, we introduced RTTE modeling, an advanced type of modeling methodology for bleeding event data in hemophilia A, to the exposure–response characterization of emicizumab. An advantage of our RTTE modeling approach over the reported NB modeling approach is that it considers the association of the continuous drug concentration–time profile itself (‘entire’ exposure variable) with the onset times of bleeding events, which allows prediction of bleeding onset as a function of dosing regimen, considering the pharmacokinetic profile differences, when concomitantly used with a PopPK model. Leveraging historical data prior to study enrollment enabled the RTTE model to characterize the natural history of bleeding onset in a non-prophylactic state, which served as the internal control for the emicizumab effect.
The RTTE model estimated the difference in bleeding hazard between patients or periods with and without prophylaxis with coagulation factor treatment to be less than the reported effect sizes [32,33,34,35,36,37, 40, 41] with high uncertainty, and therefore the estimate should be interpreted with caution. This consequence is possibly because our phase I–I/II study designs were not intended to rigorously estimate such effect sizes and/or some patients enrolled were likely to be refractory to the prophylactic treatments. Due to the large inter-individual variability in the exposure–response relationship of emicizumab, the target efficacious exposure of 45 μg/mL is not the ‘threshold’ for achieving zero ABR for every patient. Zero ABR is expected in a proportion of patients even at < 45 μg/mL, and is not expected for all patients even at ≥ 45 μg/mL. The validity of the target efficacious exposure will need to be confirmed in phase III studies in the future.
We had several points to consider for the phase III dose selection of emicizumab. First, due to its molecular structure difference from FVIII, the bleeding-prophylactic effect of emicizumab is not affected by existing FVIII inhibitors. The selection of phase III dosing regimens therefore aimed to provide comparable efficacy with prophylaxis with FVIII for patients both with and without FVIII inhibitors. Second, due to its long elimination half-life of approximately 1 month in patients, constant maintenance of high hemostatic activity with small peak–trough fluctuations is possible with QW or even less frequent dosing of emicizumab. According to the previously estimated conversion factor for the equivalent FVIII activity of emicizumab (i.e., ≥ 0.3 IU/dL per μg/mL) [18], the maintenance dose of 1.5 mg/kg QW is expected to constantly provide an equivalent FVIII activity of at least 15 IU/dL, which should minimize the risk of joint bleeding [42] and may prevent hemophilic arthropathy [43]. These characteristics will offer a novel paradigm of hemophilia A treatment which has not been achieved by existing treatments. Third, availability of multiple options in dosing regimen would meet patients’ need for dosing convenience and may thus improve their therapeutic adherence, which could in turn lead to improvements in health conditions and quality of life. The pharmacometric approach enabled investigation of previously untested, less frequent maintenance doses of 3 mg/kg Q2W and 6 mg/kg Q4W in phase III studies. These dosing regimens, with the equivalent cumulative doses to 1.5 mg/kg QW, are expected to provide similar efficacy. The same loading dose was selected irrespective of the maintenance dose for simplicity. Finally, the selected phase III dosing regimens have a 2-fold margin in cumulative dose compared with the highest previously tested dosing regimen of 3 mg/kg QW. This enables intra-patient dose up-titration from the selected dosing regimens to 3 mg/kg QW in case of suboptimal bleeding control in phase III studies.
Dose-ascending study designs, often employed for small-sized, early-phase clinical studies, do not randomize patients across dosing groups. This may cause imbalanced baseline disease characteristics due to potential biases in enrolling patients, and may therefore complicate determination of the dose–response relationship simply based on the observed data. However, in rare diseases with a limited number of patients available for study enrollment, conducting a series of adequately powered, randomized, controlled clinical studies throughout the development program, including a conventional dose-finding study, may not be feasible [44,45,46,47], especially in the presence of high unmet medical need where rapid access to new drugs is strongly desired. For dose–response characterization, non-linear mixed-effect (‘population’) modeling, which considers inter-individual variability and influential covariates, appears to be an appropriate methodology to analyze limited early-phase clinical study data [45,46,47,48,49]. Such a pharmacometric approach can enable dose selection for pivotal studies without conducting a conventional dose-finding study and will play a key role in warranting rapid development without losing scientific validity for new drugs in rare diseases where evaluable clinical efficacy endpoints are available.
Limitations to this research include the analysis results being derived from a small number of patients. In addition, the baseline disease characteristics of enrolled patients and bleeding event definition may differ across clinical studies, which may affect the consistency in efficacy results between the model-based simulations and actual observations in subsequent studies. Neither discontinuation of emicizumab treatment, modification or deviation of dosing regimens, nor development of NAbs were considered for the simulations. All the simulations for the 6 mg/kg Q4W dosing regimen assumed linear pharmacokinetics beyond the highest previously tested dose of 3 mg/kg. Of note, pediatric dose selection may require additional specific considerations and is out of the scope of this research.
5 Conclusions
A pharmacometric approach guided the selection of previously untested dosing regimens of emicizumab for phase III studies, without conducting a conventional dose-finding study in the clinical development program in hemophilia A. The conduct of phase III studies with the selected dosing regimens has been approved by health authorities. Consequently, three global phase III studies in adult and adolescent patients with or without FVIII inhibitors are currently ongoing (ClinicalTrials.gov identifiers NCT02622321, NCT02847637, and NCT03020160). This experience indicates that a pharmacometric analysis leveraging early-phase clinical study data can provide a substitute for a conventional dose-finding study and will streamline the drug development process in rare diseases.
References
Mannucci PM, Tuddenham EG. The hemophilias–from royal genes to gene therapy. N Engl J Med. 2001;344:1773–9.
Geraghty S, Dunkley T, Harrington C, Lindvall K, Maahs J, Sek J. Practice patterns in haemophilia A therapy—global progress towards optimal care. Haemophilia. 2006;12:75–81.
Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935–9.
Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–47.
Collins PW, Fischer K, Morfini M, et al. Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia. 2011;17:2–10.
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44.
Collins P, Chalmers E, Chowdary P, et al. The use of enhanced half-life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia. 2016;22:487–98.
De Moerloose P, Urbancik W, Van Den Berg HM, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14:931–8.
du Treil S, Rice J, Leissinger CA. Quantifying adherence to treatment and its relationship to quality of life in a well-characterized haemophilia population. Haemophilia. 2007;13:493–501.
Fischer K, Lewandowski D, Marijke van den Berg H, Janssen MP. Validity of assessing inhibitor development in haemophilia PUPs using registry data: the EUHASS project. Haemophilia. 2012;18:e241–6.
Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054–64.
Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–4.
Sampei Z, Igawa T, Soeda T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8:e57479.
Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117:1348–57.
Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12:206–13.
Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody ACE910 prevents joint bleeds in a long-term primate model of acquired hemophilia A. Blood. 2014;124:3165–71.
Uchida N, Sambe T, Yoneyama K, et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633–41.
Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374:2044–53.
Shima M, Hanabusa H, Taki M, et al. Long-term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Adv. 2017;1:1891–9.
Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–32.
Björkman S, Folkesson A, Jönsson S. Pharmacokinetics and dose requirements of factor VIII over the age range 3–74 years: a population analysis based on 50 patients with long-term prophylactic treatment for haemophilia A. Eur J Clin Pharmacol. 2009;65:989–98.
Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56:4927–36.
Boeckmann AJ, Sheiner LB, Beal SL. NONMEM users guide—part VIII: help guide. Ellicott City: ICON Development Solutions; 2011.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–51.
Thai HT, Mentré F, Holford NH, Veyrat-Follet C, Comets E. Evaluation of bootstrap methods for estimating uncertainty of parameters in nonlinear mixed-effects models: a simulation study in population pharmacokinetics. J Pharmacokinet Pharmacodyn. 2014;41:15–33.
Cox EH, Veyrat-Follet C, Beal SL, Fuseau E, Kenkare S, Sheiner LB. A population pharmacokinetic-pharmacodynamic analysis of repeated measures time-to-event pharmacodynamic responses: the antiemetic effect of ondansetron. J Pharmacokinet Biopharm. 1999;27:625–44.
Plan EL, Ma G, Någård M, Jensen J, Karlsson MO. Transient lower esophageal sphincter relaxation pharmacokinetic-pharmacodynamic modeling: count model and repeated time-to-event model. J Pharmacol Exp Ther. 2011;339:878–85.
Holford NH, Sheiner LB. Kinetics of pharmacologic response. Pharmacol Ther. 1982;16:143–66.
Tuk B, van Oostenbruggen MF, Herben VM, Mandema JW, Danhof M. Characterization of the pharmacodynamic interaction between parent drug and active metabolite in vivo: midazolam and alpha-OH-midazolam. J Pharmacol Exp Ther. 1999;289:1067–74.
Earp J, Krzyzanski W, Chakraborty A, Zamacona MK, Jusko WJ. Assessment of drug interactions relevant to pharmacodynamic indirect response models. J Pharmacokinet Pharmacodyn. 2004;31:345–80.
Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28:171–92.
Manco-Johnson MJ, Kempton CL, Reding MT, et al. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11:1119–27.
Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on-demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359–67.
Kavakli K, Yang R, Rusen L, et al. Prophylaxis vs. on-demand treatment with BAY 81-8973, a full-length plasma protein-free recombinant factor VIII product: results from a randomized trial (LEOPOLD II). J Thromb Haemost. 2015;13:360–9.
Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian™1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19:691–7.
Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126:1078–85.
Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317–25.
Collins PW, Blanchette VS, Fischer K, et al. Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost. 2009;7:413–20.
Valentino LA, Pipe SW, Collins PW, et al. Association of peak factor VIII levels and area under the curve with bleeding in patients with haemophilia A on every third day pharmacokinetic-guided prophylaxis. Haemophilia. 2016;22:514–20.
Antunes SV, Tangada S, Stasyshyn O, et al. Randomized comparison of prophylaxis and on-demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitors. Haemophilia. 2014;20:65–72.
Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5:1904–13.
den Uijl IE, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17:41–4.
Ling M, Heysen JP, Duncan EM, Rodgers SE, Lloyd JV. High incidence of ankle arthropathy in mild and moderate haemophilia A. Thromb Haemost. 2011;105:261–8.
Gobburu J, Pastoor D. Drugs against rare diseases: are the regulatory standards higher? Clin Pharmacol Ther. 2016;100:322–3.
Bashaw ED, Huang SM, Coté TR, et al. Clinical pharmacology as a cornerstone of orphan drug development. Nat Rev Drug Discov. 2011;10:795–6.
Lesko LJ. Quantitative analysis to guide orphan drug development. Clin Pharmacol Ther. 2012;92:258–61.
Nony P, Kurbatova P, Bajard A, et al. A methodological framework for drug development in rare diseases. Orphanet J Rare Dis. 2014;9:164.
Bhattacharya I, Manukyan Z, Chan P, Harnisch L, Heatherington A. Making every subject count: a case study of drug development path for medication in a pediatric rare disease. Clin Pharmacol Ther. 2016;100:330–2.
Aarons L, Karlsson MO, Mentré F, et al. Role of modelling and simulation in Phase I drug development. Eur J Pharm Sci. 2001;13:115–22.
Acknowledgements
The authors wish to thank all the patients and their families and the volunteers who participated in the clinical studies, investigators and staff at the study institutions, and colleagues at Chugai Pharmaceutical Co., Ltd. (Chugai) and F. Hoffmann-La Roche Ltd. (Roche), especially Tomohisa Saito and Hiroki Yoshida for data analytical support, and Naoki Fukazawa for valuable comments.
Author information
Authors and Affiliations
Contributions
K.Y. and C.S. designed the research. K.Y. analyzed the data and wrote the manuscript. All authors interpreted the data.
Corresponding author
Ethics declarations
Funding
This research was sponsored by Chugai and Roche. Open access publication of this manuscript was sponsored by Chugai and Roche.
Conflict of interest
K.Y., N.K., R.K., S.I., and T.K. are employees of Chugai. C.S. is an employee of F. Hoffmann-La Roche AG. G.G.L. is an employee of Genentech, Inc. G.G.L. and T.K. hold stock/stock options of Roche and Chugai, respectively. M.S. has received a research grant from Chugai, and consulting honoraria from Chugai and Roche. K.Y., C.S., and M.S. are inventors of the patents relating to anti-FIXa/X bispecific antibodies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yoneyama, K., Schmitt, C., Kotani, N. et al. A Pharmacometric Approach to Substitute for a Conventional Dose-Finding Study in Rare Diseases: Example of Phase III Dose Selection for Emicizumab in Hemophilia A. Clin Pharmacokinet 57, 1123–1134 (2018). https://doi.org/10.1007/s40262-017-0616-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0616-3