
Abstract
The most recent comprehensive reviews on the population pharmacokinetics of mycophenolic acid (MPA) were published in 2014. Since then, several population pharmacokinetic studies on MPA have been published. The majority of literature is still focused on the kidney transplant population, although studies have also been conducted in liver and lung transplantation, autoimmune diseases, and hematopoietic stem cell transplant. While the majority of the model building is still based on parametric non-linear mixed-effects modeling, recent studies suggest the suitability of other methodologies. Additionally, instead of just focusing on pharmacokinetic modeling, a trend toward describing the relationships between pharmacokinetic and pharmacodynamic parameters is observed. Given the importance of enterohepatic recirculation (EHR) in the pharmacokinetics of MPA, more authors have attempted to characterize this process in their models. Overall, the recent models have become more sophisticated and incorporate EHR, pharmacodynamic relationships, and metabolites while maintaining many of the population values and covariates identified previously. However, the number of MPA population pharmacokinetic models describing the enteric-coated formulation of MPA (EC-MPA) is still limited. Given the increasing use of EC-MPA, more studies are needed to fill this literature gap. In addition, few studies are yet available characterizing free MPA concentration or MPA metabolites. Given the extensive protein binding, low to intermediate extraction, and intrinsic clearance characteristics of MPA in humans, including these variables would improve the population structural models.
Similar content being viewed by others

Many population models are built on mixed patient populations (combining more than one transplant type, stem cell transplant, or autoimmune disease) that can assess the influence of specific patient type (as covariates) on mycophenolic acid (MPA) pharmacokinetics within the same model. |
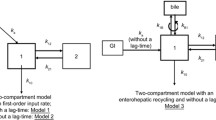
Non-linear mixed-effects modeling using first-order conditional estimation with or without interaction is still the gold standard used in most studies. Most studies employ a two-compartment model (central, peripheral compartment with various types of absorption or elimination) to characterize the final structural model. |
The final model parameters reported in recent population pharmacokinetic studies are in general agreement with those reported previously and summarized in aforementioned reviews, although various novel findings have been identified. |
To fill the literature gap, future population pharmacokinetic studies are needed to characterize enteric-coated MPA, free MPA concentration, enterohepatic recirculation, and MPA metabolites. |
1 Introduction
Mycophenolic acid (MPA) is an immunosuppressant commonly used in solid organ (kidney, liver, and lung) transplantation [1, 2], autoimmune diseases [3], and hematopoietic stem cell transplants [4]. It exerts its immunosuppression effects by selectively reducing guanine nucleotide synthesis via the reversible inhibition of inosine monophosphate dehydrogenase (IMPDH) [5]. MPA is almost always used in combination with additional immunosuppressants (such as calcineurin inhibitors plus or minus corticosteroids), which results in complex pharmacokinetics as a direct consequence of intrinsic (i.e. patient and disease) and extrinsic (e.g. concurrent coadministered medications) covariates. A fixed-dose regimen is still the current convention in MPA dosing because the therapeutic range [using area under the curve (AUC) as the optimal surrogate marker] was established only in kidney transplant subjects under specific conditions (with coadministered drugs that may not be in common practice today), making the recommendation of concentration-based MPA dosing not universally accepted among clinicians [6].
Various factors are known to contribute to the large variabilities observed in MPA pharmacokinetics in humans. MPA is extensively absorbed from the gastrointestinal tract and is almost completely bound to serum albumin [7]. The absorption characteristics of MPA differ significantly between the two available oral dosage forms [mycophenolate mofetil (MMF) and the enteric-coated salt, mycophenolate sodium (EC-MPA)]. For example, the time to reach the maximum concentration, trough concentration, and drug exposure have all been demonstrated to be different between the two formulations [1], and, as such, they are not considered bioequivalent due to their highly variable pharmacokinetics. For these reasons, even though the amount of active MPA (739 mg) released from 1 g of MMF [2] is approximately equivalent to 720 mg of EC-MPA, the two formulations are not interchangeable. Because only free MPA is subject to metabolism, and total MPA concentration may fluctuate in the absence of a change in free concentration, protein-binding displacement (e.g. secondary to hypoalbuminemia or concurrent albumin-binding drugs) contributes to altered total, but not free, MPA clearance and makes MPA therapeutic drug monitoring difficult. Although it would be ideal to characterize MPA pharmacokinetics using free drug concentrations, that is still rarely done today.
The processes of intrinsic clearance and drug transport can also contribute to the variability of MPA pharmacokinetics in human subjects. MPA is primarily conjugated by UDP-glucuronosyltransferase (UGT) enzymes in the production of the major, inactive, MPA-glucuronide (MPAG) and the relatively minor, but active, MPA-acyl-glucuronide (AcMPAG) [1]. MPA glucuronides are excreted unchanged in the urine, but also undergo subsequent enterohepatic recirculation (EHR), mediated via multidrug-resistant protein-2 (MRP-2), in the re-regeneration of a significant amount of MPA. Variability in the EHR process can result in multiple peaks in the plasma drug concentration-time curves of MPA, which are difficult to predict, yet this crucial process is not always characterized in MPA pharmacokinetic modeling. In addition, despite many potential drug–drug or drug–gene interactions involved in the intrinsic clearance (UGT) or transport (MRP-2, e.g. cyclosporine) of MPA, the majority of MPA pharmacokinetic studies to date have focused only on the parent compound, potentially negating the important roles of metabolism, metabolites, and the EHR process in the pharmacokinetics of MPA.
Disease state-specific pharmacokinetic parameters, intersubject variability, interoccasion variabilities, and the influence of covariates (intrinsic or extrinsic) are best characterized by population pharmacokinetic modeling [8, 9]. Several excellent reviews have been published on the population pharmacokinetics of MPA in specific patient populations: solid organ transplant [2, 10], autoimmune disease [3, 10], and stem cell transplant [4] (the latter a qualitative review). Mechanism-focused reviews on EHR [10], and a more generalized review [11], are also available and bring the literature summary (with quantitative pharmacokinetic data) up-to-date to late 2013. However, since that time, several population pharmacokinetic studies on MPA have been published that warrant an updated analysis. This systematic review aims to objectively evaluate and summarize the more ‘recent data’ in order to provide an updated summary on the overall landscape for the population pharmacokinetics of MPA for all relevant indications. Our definition of ‘recent data’ is as follows: primary papers on MPA population pharmacokinetic modeling for any indication since September 2013 (i.e. to our knowledge, the latest quantitative pharmacokinetic review on the subject matter) or any earlier papers that have not been summarized in a quantitative manner in the aforementioned reviews.
2 Methodology
The following databases, from inception to June 2017, were searched (PubMed, EMBASE, and Google Scholar) using combinations of search terms (mycophenolate, mycophenolic acid, mycophenolic acid sodium, Cellcept, Myfortic, pharmacokinetics, pharmacodynamics, population pharmacokinetics, non-linear mixed-effects modeling, solid organ transplant, kidney, liver, lung, autoimmune disease, hematopoietic stem cell transplant, modeling). No search limits (other than English language only) were applied. The reference lists of identified articles were also manually searched to ensure comprehensiveness. Only population pharmacokinetic analyses involving actual patients were included in this review (i.e. those involving healthy subjects were excluded for lack of clinical utility). Data already summarized in the aforementioned reviews in the Introduction section were also excluded from this paper (see our definition of ‘recent data’). Data are summarized in Table 1 based on indication, in chronological order. Some studies may appear more than once in the table because the categorization is based on patient population and a few studies enrolled more than one patient population. The (1) general population characteristics, (2) model building details, and (3) final model parameters (i.e. average population pharmacokinetic values, significant covariates, intersubject variability, and residual error) are systematically summarized, in addition to limitations and novelty, for each patient population.
3 Results
3.1 Kidney Transplant
The majority of the population pharmacokinetic literature on MPA has been, and is still, based on the kidney transplant population (Table 1). Recent MPA population pharmacokinetic data in pediatric [12,13,14] and adult subjects [15,16,17,18] are summarized in Table 1. In terms of the general population characteristics, with the exception of Han et al. [19], Musuamba et al. [16], and Veliĉković-Radovanović et al. [18] who used EC-MPA, all studies administered MMF. Many population models were built on mixed patient populations (combining more than one transplant type, stem cell transplant, or autoimmune disease) [12, 15, 17], thus potentially limiting the specificity of their findings to kidney transplant subjects. On the other hand, these mixed population models allow the assessment of the influence of specific patient types (as covariates) on MPA pharmacokinetics (e.g. the characterization of apparent oral clearance [CL/F] values in each patient type in the study by de Winter et al. [15]) within the same model. Overall, kidney transplant patients enrolled in these studies received concurrent calcineurin inhibitors (tacrolimus or cyclosporine in the study population) and a corticosteroid, but the information provided on other coadministered drugs was minimal, potentially limiting the identification of drug covariates in these models (Table 1).
With respect to model building, non-linear mixed-effects modeling using first-order conditional estimation (FOCE) with or without interaction is still the gold standard used in all studies (Table 1). Most studies only focused on population pharmacokinetic structural model building; however, Premaud et al. [13] specifically compared non-linear mixed-effects modeling (parametric) against the adaptive grid (non-parametric) approach and generated Bayesian forecasting equations for the prediction of MPA pharmacokinetics. Dong et al. [14] constructed a population pharmacokinetic–pharmacodynamic model to better characterize the relationship between MPA concentrations and IMPDH activities. To determine the validity of these models, all studies (with the exception of Veliĉković-Radovanović et al. [18]) utilized internal validation (bootstrapping) in conjunction with visual predictive checks (Table 1) as the primary approach to model qualification. The use of internal validation is common, but less ideal compared to a randomly assigned external (independent) validation/test group [8, 9], such as that used by Veliĉković-Radovanović et al. [18]. All studies employed a two-compartment model (central, peripheral compartment with various types of absorption or elimination) to characterize the final structural model (Table 1). This is consistent with the many population pharmacokinetic two-compartment models already published in kidney transplant patients [2, 10]. Unfortunately, none of these studies characterized metabolite concentrations or had sufficient sampling points to determine the EHR of MPA, which may have simplified the model building process but also likely limited the overall accuracy/precision of the final structural model.
The final model parameters reported by these population pharmacokinetic studies (Table 1) are in general agreement with those reported previously and summarized in the aforementioned reviews. Various novel findings are identified as follows: Zeng et al. [12] constructed a population model describing both intravenous/oral administration of MMF in a mixed pediatric population model; de Winter et al. [15] provided evidence that mean MPA clearance values varied significantly between adult kidney, hematopoietic stem cell, and autoimmune disease patients; Premaud et al. [13] demonstrated that the non-parametric approaches to population pharmacokinetic model building yielded more accurate and precise Bayesian forecasting equations (for MPA exposure) than the current gold-standard approach (non-linear mixed-effects modeling); Musuamba et al. [16] provided data supporting a significant effect of the formulation (EC-MPA vs. MMF) on the pharmacokinetics of MPA; Han et al. [19] added to the limited literature on the population pharmacokinetics of MPA from the administration of EC-MPA; Dong et al. [14] constructed one of the first pharmacokinetic-pharmacodynamic population models in the pediatric kidney transplant population; and Veliĉković-Radovanović et al. [18] provided evidence indicating an effect from nifedipine coadministration on MPA clearance (Table 1). The covariates identified in these studies are also in general agreement with those already published previously [e.g. that albumin, cyclosporine, body weight (in pediatric patients), MRP-2 genetic polymorphism, SLCO1B1 polymorphism, and UGT1A9 polymorphism affect the clearance of MPA]. The fact that nifedipine is identified as a covariate in the final structural model in the study by Veliĉković-Radovanović et al. [18] would require further investigation from a mechanistic (i.e. metabolism-related) point of view as the nifedipine–MPA interaction is not commonly recognized in clinical practice today. The apparent lack of newly identified (other than nifedipine) covariates in the kidney transplant population in these publications might be due to the small covariate selection and small sample sizes (i.e. thus, lack of power). Finally, the interpatient and residual variabilities are also in line with those reported previously. The generally large variability observed (Table 1) further supports the already-established notion of substantial variability (explained and unexplained) in the pharmacokinetics of MPA (Table 1).
3.2 Liver Transplant
Similar to the existing data, more recently published population pharmacokinetic literature of MPA remains scarce in the liver transplant population (Table 1). Specifically, MPA population pharmacokinetic data in pediatric [12] and adult subjects [20] are summarized in Table 1. In terms of the general population characteristics, all studies administered MMF, preventing the generalizability of these population models to patients taking EC-MPA. As described in the kidney transplant population, some models were built on mixed patient populations (combining more than one transplant type, stem cell transplant, or autoimmune disease) [12], thus potentially limiting the applicability of their findings to the liver transplant subjects. Overall, liver transplant patients enrolled in these studies received concurrent calcineurin inhibitors (tacrolimus or cyclosporine) and a corticosteroid, but corticosteroid use was not specified in the pediatric patients studied by Zeng et al. [12]. Again, the information provided on other coadministered drugs was minimal, potentially limiting the identification of drug covariates in these models (Table 1).
With respect to model building, non-linear mixed-effects modeling using FOCE with interaction and the iterative two-stage model with Bayesian determination was used (Table 1). Although the studies focused on structural model building, Langers et al. [20] also generated a Bayesian forecasting equation for the prediction of MPA pharmacokinetics. To determine the validity of these models, Zeng et al. [12] utilized internal validation (bootstrapping) in conjunction with visual predictive checks as the primary approach to model qualification, but the validation method by Langers et al. [20] was not clearly reported. Both studies employed a two-compartment model (central, peripheral compartment with first-order elimination or absorption) to characterize the final structural model (Table 1). This is consistent with the existing population pharmacokinetic two-compartment models already published in liver transplant patients [2, 10]. Similar to the kidney transplant population, neither of these studies characterized metabolite concentrations or had sufficient sampling points to determine the EHR of MPA.
The final model parameters reported by these two population pharmacokinetic studies (Table 1) are in general agreement with that reported previously and summarized in the aforementioned reviews. We identified the following novel findings: Zeng et al. [12] constructed a population model describing both intravenous/oral administration of MMF in a mixed pediatric population model, while Langers et al. [20] provided a new limited sampling equation based on Bayesian modeling in adult liver transplant subjects (Table 1). The covariates identified in these studies are also in general agreement with that already published previously [e.g. that albumin, creatinine clearance, cyclosporine, and body weight (in pediatric patients) affect the clearance of MPA]. However, Langers et al. [20] did not find a significant correlation between the type of coadministered immunosuppressant (e.g. tacrolimus vs. cyclosporine vs. sirolimus) and the pharmacokinetics of MPA. This is in contrast to the established fact that cyclosporine, but not tacrolimus, has a significant effect on MPA clearance (e.g. [21, 22]). The discrepancy from Langers et al. might be secondary to the relatively small sample size (N = 57) and the fact that only two covariates (albumin and creatinine clearance) were subjected to the model building process. Finally, where available, the interpatient and residual variabilities are also in line with those reported previously. The generally large variability observed (Table 1) further supports the already-established notion of substantial variability (explained and unexplained) in the pharmacokinetics of MPA (Table 1).
3.3 Lung Transplant
Similar to the liver transplant patients, population pharmacokinetic literature of MPA remains scarce in the lung transplant population (Table 1). Only data from adult subjects are available (summarized in Table 1). In terms of the general population characteristics, all studies administered MMF, preventing the generalizability of these population models to patients taking EC-MPA. The population model from de Winter et al. [17] was built on mixed patient populations (combining kidney and lung transplant recipients). On the other hand, the two studies by Wang et al. [23, 24] specifically compared the pharmacokinetics between patients with or without cystic fibrosis. Overall, lung transplant patients enrolled in these studies received concurrent calcineurin inhibitors (tacrolimus or cyclosporine) and a corticosteroid, but corticosteroid use was not specified in the mixed population studied reported by de Winter et al. [17]. Similar to other patient populations described in this review, the information provided on other coadministered drugs was minimal, potentially limiting the identification of drug covariates in these models (Table 1).
With respect to model building, non-linear mixed-effects modeling using FOCE with or without interaction is still the gold standard used by all studies (Table 1). All studies focused on population pharmacokinetic structural model building, but de Winter et al. [17] generated a Bayesian forecasting equation for the prediction of MPA pharmacokinetics in lung transplant patients. To determine the validity of these models, all three studies utilized internal validation (bootstrapping) in conjunction with visual predictive checks (Table 1) as the primary approach to model qualification. In the study by de Winter et al. [17], an external validation cohort including only lung transplant patients was utilized to determine the predictive performance of the generated Bayesian limited sampling equation. All studies employed a two-compartment model (central, peripheral compartment with various types of absorption or elimination) to characterize the final structural model (Table 1). The novel model developed by Wang et al. [24] was more complex, incorporating an MPAG compartment to further characterize EHR. These two-compartment models are generally consistent with the existing population pharmacokinetic two-compartment models already published in lung transplant patients [2, 10].
The final model parameters reported by these population pharmacokinetic studies (Table 1) are also in general agreement with those reported previously and summarized in the aforementioned reviews. The novel findings are identified as follows: de Winter et al. [17] generated a novel Bayesian estimator using a mix of lung and kidney transplant patients that was suitable for estimating MPA pharmacokinetic parameters in lung transplant patients, while Wang et al. [23, 24] constructed a population model suitable for describing the pharmacokinetics (with EHR) of lung transplant patients with or without cystic fibrosis (Table 1). The covariates identified in these studies are consistent with the existing literature that cystic fibrosis affects the clearance of MPA. Finally, the interpatient and residual variabilities are also inline with those reported previously. The generally large variability observed (Table 1) further supports the already-established notion of substantial variability (explained and unexplained) in the pharmacokinetics of MPA (Table 1).
3.4 Autoimmune Diseases
Recent MPA population pharmacokinetic data in pediatric patients with idiopathic nephrotic syndrome [25] or systemic lupus erythematosus [26], and adult subjects with various types of autoimmune disease [15, 27], are summarized in Table 1. In terms of the general population characteristics, all studies administered MMF, which does not alleviate the shortage of data on the enteric-coated dosage form. The population model from de Winter et al. [15] was built on mixed populations (kidney transplant, hematopoietic stem cell transplant, and autoimmune disease), whereas Saint-Marcoux et al. [25] and Abd Rahman et al. [27] focused on idiopathic nephrotic syndrome and lupus nephritis, respectively. Overall, autoimmune patients enrolled in these studies received concurrent corticosteroids (except for the study by Woillard et al. [26] in which concurrent medications were not identified). Except in the study described by Abd Rahman et al. [27], the other patient populations received no calcineurin inhibitors. Again, the information provided on other coadministered drugs was minimal, potentially limiting the identification of drug covariates in these models (Table 1).
With respect to model building, non-linear mixed-effects modeling using FOCE with or without interaction was used by de Winter et al. [15] and Abd Rahman et al. [27], whereas Saint-Marcoux et al. [25] and Woillard et al. [26] utilized the iterative two-stage modeling approach. All studies focused on population pharmacokinetic structural model building, but Saint-Marcoux et al. [25] and Woillard et al. [26] generated a Bayesian forecasting equation for the prediction of MPA pharmacokinetics. To determine the validity of these models, all studies utilized internal validation (bootstrapping) [except Woillard et al.] in conjunction with visual predictive checks (Table 1) as the primary approach to model qualification. In addition, Saint-Marcoux et al. [25] incorporated an external validation subset consisting of 15 pediatric patients with idiopathic nephrotic syndrome. With the exception of Saint-Marcoux et al. [25] and Woillard et al. [26], who described a one-compartment model, the other studies utilized a two-compartment set-up (central, peripheral compartment with various types of absorption or elimination) to characterize the final structural model (Table 1). Moreover, additional model parameters were generated by Saint-Marcoux et al. [25] and Abd Rahman et al. [27], with the latter also determining MPAG concentration to describe the EHR process. On the other hand, despite the intense sampling data used by Woillard et al. [26], they did not attempt to characterize or model EHR.
The final model parameters reported by these population pharmacokinetic studies (Table 1) are in general agreement with those reported previously. Various novel findings are identified as follows: de Winter et al. [15] provided evidence that mean MPA clearance values varied significantly between adult kidney, hematopoietic stem cell, and autoimmune disease patients; Saint-Marcoux et al. [25] developed a Bayesian estimator for the determination of MPA AUC in pediatric patients with idiopathic nephrotic syndrome; Woillard et al. [26] provided the first Bayesian estimator for the calculation of MPA exposure in pediatric lupus patients; and Abd Rahman et al. [27] characterized the population pharmacokinetics of adult lupus patients using a more comprehensive approach, including metabolite and EHR data (Table 1). The covariates identified in these studies are also in general agreement with those already published (e.g. albumin, cyclosporine affects the clearance of MPA). Abd Rahman et al. [27] provided the novel findings that renal function affects the CL/F of MPAG, and concrete evidence that cyclosporine use does affect MPAG EHR (confirming the data only observed with MPA alone in other studies (e.g. [28]). Again, the generally large variability observed (Table 1) supports the already-established notion of substantial variability (explained and unexplained) in the pharmacokinetics of MPA (Table 1).
3.5 Hematopoietic Stem Cell Transplant
A limited number of studies have been identified describing the population pharmacokinetics of MPA in stem cell transplant patients (Table 1). Because Zeng et al. [12] and de Winter et al. [15] utilized mixed patient populations, these two studies have already been described in other sections in this review. Li et al. [29] developed a population pharmacokinetic-pharmacodynamic model describing the concentration-effect relationship of MPA in adult non-myeloablative allogenic hematopoietic stem cell transplant patients. Model building utilized a non-linear mixed-effects approach with the Monte Carlo ‘important sampling expectation maximization’. The authors included total MPA, free MPA, MPAG, and IMPDH activity in their final structural model, which was best described as a two-compartment model with first-order elimination/absorption. MPAG was characterized by an additional compartment (with first-order elimination) and the pharmacodynamic relationship was determined by the maximal effect (E max) model (Table 1). This novel pharmacokinetic–pharmacodynamic model was validated internally and externally, and the typical covariates (serum creatinine, cyclosporine affecting the pharmacokinetics of MPA or MPAG) were identified. Some of the population parameters generated in this study (with the exception of the new parameters identified) are comparable with those already summarized in the literature, indicating the validity of their approach.
4 Conclusions and Future Directions
This article brings the review literature of MPA population pharmacokinetics up to the present day. It summarizes the relevant population models developed in all patient populations for which MPA is indicated (Table 1). In summary, while the majority of the model building is still based on parametric non-linear mixed-effects modeling, studies have become available describing (and suggesting the suitability of) other methodologies (Table 1). In addition, instead of just focusing on pharmacokinetic modeling, a trend toward describing the relationships between pharmacokinetic and pharmacodynamic parameters is observed. Furthermore, given the importance of EHR in the pharmacokinetics of MPA, more and more authors have attempted to characterize this process in their models. However, due to the relatively more intense sampling requirements needed to characterize EHR, this aspect of the modeling may not be feasible in many clinical settings. Overall, the recent models have become more sophisticated (incorporating EHR, pharmacodynamic relationships, and metabolites) while maintaining many of the population values and covariates identified previously. On the other hand, the number of MPA population pharmacokinetic models describing the EC-MPA is still limited. Given the increasing trend of using EC-MPA in patients, more studies are urgently needed to fill this important literature gap. In addition, few studies are yet available characterizing free MPA concentration or MPA metabolites. Given the extensive protein binding, low to intermediate extraction, and intrinsic clearance characteristics of MPA in humans, including these variables would certainly improve the population structural models. In addition, the clinical relevance of the developed models would also require further investigations. For example, the benefits of any population Bayesian predictive equations would require further clinical trials in order to test the hypothesis that their utilization can result in adequate changes to clinical outcomes (e.g. graft rejection, neutropenia, etc.). Finally, universal standards and best practices for population pharmacokinetic model building and validation (e.g. as proposed by the Standards and Best Practices Committee in the International Society of Pharmacometrics (http://www.go-isop.org/standards-best-practices-committee) should be followed to ensure future modeling activities for MPA are consistent with the established guidelines.
References
Staatz CE, Tett SE. Pharmacology and toxicology of mycophenolate in organ transplant recipients: an update. Arch Toxicol. 2014;88(7):1351–89.
Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin Pharmacokinet. 2007;46(1):13–58.
Abd Rahman AN, Tett SE, Staatz CE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in patients with autoimmune disease. Clin Pharmacokinet. 2013;52(5):303–31.
Zhang D, Chow DS. Clinical pharmacokinetics of mycophenolic acid in hematopoietic stem cell transplantation recipients. Eur J Drug Metab Pharmacokinet. 2017;42(2):183–9.
Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl 1):s2–8.
Kiang TK, Ensom MH. Therapeutic drug monitoring of mycophenolate in adult solid organ transplant patients: an update. Expert Opin Drug Metab Toxicol. 2016;12(5):545–53.
Tett SE, Saint-Marcoux F, Staatz CE, Brunet M, Vinks AA, Miura M, et al. Mycophenolate, clinical pharmacokinetics, formulations, and methods for assessing drug exposure. Transplant Rev (Orlando). 2011;25(2):47–57.
Kiang TK, Sherwin CM, Spigarelli MG, Ensom MH. Fundamentals of population pharmacokinetic modelling: modelling and software. Clin Pharmacokinet. 2012;51(8):515–25.
Sherwin CM, Kiang TK, Spigarelli MG, Ensom MH. Fundamentals of population pharmacokinetic modelling: validation methods. Clin Pharmacokinet. 2012;51(9):573–90.
Sherwin CM, Fukuda T, Brunner HI, Goebel J, Vinks AA. The evolution of population pharmacokinetic models to describe the enterohepatic recycling of mycophenolic acid in solid organ transplantation and autoimmune disease. Clin Pharmacokinet. 2011;50(1):1–24.
Dong M, Fukuda T, Vinks AA. Optimization of mycophenolic acid therapy using clinical pharmacometrics. Drug Metab Pharmacokinet. 2014;29(1):4–11.
Zeng L, Blair EY, Nath CE, Shaw PJ, Earl JW, Stephen K, et al. Population pharmacokinetics of mycophenolic acid in children and young people undergoing blood or marrow and solid organ transplantation. Br J Clin Pharmacol. 2010;70(4):567–79.
Premaud A, Weber LT, Tonshoff B, Armstrong VW, Oellerich M, Urien S, et al. Population pharmacokinetics of mycophenolic acid in pediatric renal transplant patients using parametric and nonparametric approaches. Pharmacol Res. 2011;63(3):216–24.
Dong M, Fukuda T, Cox S, de Vries MT, Hooper DK, Goebel J, et al. Population pharmacokinetic–pharmacodynamic modelling of mycophenolic acid in paediatric renal transplant recipients in the early post-transplant period. Br J Clin Pharmacol. 2014;78(5):1102–12.
de Winter BC, Mathot RA, Sombogaard F, Neumann I, van Hest RM, Doorduijn JK, et al. Differences in clearance of mycophenolic acid among renal transplant recipients, hematopoietic stem cell transplant recipients, and patients with autoimmune disease. Ther Drug Monit. 2010;32(5):606–14.
Musuamba FT, Mourad M, Haufroid V, Demeyer M, Capron A, Delattre IK, et al. A simultaneous d-optimal designed study for population pharmacokinetic analyses of mycophenolic acid and tacrolimus early after renal transplantation. J Clin Pharmacol. 2012;52(12):1833–43.
de Winter BC, Monchaud C, Premaud A, Pison C, Kessler R, Reynaud-Gaubert M, et al. Bayesian estimation of mycophenolate mofetil in lung transplantation, using a population pharmacokinetic model developed in kidney and lung transplant recipients. Clin Pharmacokinet. 2012;51(1):29–39.
Veliĉković-Radovanović RM, Jankovic SM, Milovanovic JR, Catic-Dordevic AK, Spasic AA, Stefanovic NZ, et al. Variability of mycophenolic acid elimination in the renal transplant recipients–population pharmacokinetic approach. Ren Fail. 2015;37(4):652–8.
Han N, Yun HY, Kim IW, Oh YJ, Kim YS, Oh JM. Population pharmacogenetic pharmacokinetic modeling for flip-flop phenomenon of enteric-coated mycophenolate sodium in kidney transplant recipients. Eur J Clin Pharmacol. 2014;70(10):1211–9.
Langers P, Press RR, Inderson A, Cremers SC, den Hartigh J, Baranski AG, et al. Limited sampling model for advanced mycophenolic acid therapeutic drug monitoring after liver transplantation. Ther Drug Monit. 2014;36(2):141–7.
Filler G, Zimmering M, Mai I. Pharmacokinetics of mycophenolate mofetil are influenced by concomitant immunosuppression. Pediatr Nephrol. 2000;14(2):100–4.
Pou L, Brunet M, Cantarell C, Vidal E, Oppenheimer F, Monforte V, et al. Mycophenolic acid plasma concentrations: influence of comedication. Ther Drug Monit. 2001;23(1):35–8.
Wang XX, Feng MR, Nguyen H, Smith DE, Cibrik DM, Park JM. Population pharmacokinetics of mycophenolic acid in lung transplant recipients with and without cystic fibrosis. Eur J Clin Pharmacol. 2015;71(6):673–9.
Wang XX, Liu W, Zheng T, Park JM, Smith DE, Feng MR. Population pharmacokinetics of mycophenolic acid and its glucuronide metabolite in lung transplant recipients with and without cystic fibrosis. Xenobiotica. 2017;47(8):697–704.
Saint-Marcoux F, Guigonis V, Decramer S, Gandia P, Ranchin B, Parant F, et al. Development of a Bayesian estimator for the therapeutic drug monitoring of mycophenolate mofetil in children with idiopathic nephrotic syndrome. Pharmacol Res. 2011;63(5):423–31.
Woillard JB, Bader-Meunier B, Salomon R, Ranchin B, Decramer S, Fischbach M, et al. Pharmacokinetics of mycophenolate mofetil in children with lupus and clinical findings in favour of therapeutic drug monitoring. Br J Clin Pharmacol. 2014;78(4):867–76.
Abd Rahman AN, Tett SE, Abdul Gafor HA, McWhinney BC, Staatz CE. Development of improved dosing regimens for mycophenolate mofetil based on population pharmacokinetic analyses in adults with lupus nephritis. Eur J Drug Metab Pharmacokinet. doi:10.1007/s13318-017-0420-3.
Kuypers DR, Ekberg H, Grinyo J, Nashan B, Vincenti F, Snell P, et al. Mycophenolic acid exposure after administration of mycophenolate mofetil in the presence and absence of cyclosporin in renal transplant recipients. Clin Pharmacokinet. 2009;48(5):329–41.
Li H, Mager DE, Sandmaier BM, Storer BE, Boeckh MJ, Bemer MJ, et al. Pharmacokinetic and pharmacodynamic analysis of inosine monophosphate dehydrogenase activity in hematopoietic cell transplantation recipients treated with mycophenolate mofetil. Biol Blood Marrow Transplant. 2014;20(8):1121–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Tony K. L. Kiang and Mary H. H. Ensom have no conflicts of interests to declare.
Funding
No funding was received for the preparation of this article.
Rights and permissions
About this article

Cite this article
Kiang, T.K.L., Ensom, M.H.H. Population Pharmacokinetics of Mycophenolic Acid: An Update. Clin Pharmacokinet 57, 547–558 (2018). https://doi.org/10.1007/s40262-017-0593-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0593-6