
Abstract
Background and Objectives
Phenylephrine HCl 10 mg has been used as a nasal decongestant for over 50 years, yet only limited pharmacokinetic and metabolic data are available. The purpose of this study was to evaluate single-dose pharmacokinetics and safety of phenylephrine HCl 10, 20, and 30 mg and to assess cardiovascular tolerability compared with baseline and placebo in healthy volunteers.
Methods
Twenty-eight adults were enrolled in this randomized, double-blind, placebo-controlled, single-dose, four-treatment crossover study. Subjects remained housed for 6 days to permit time-matched, serial measurements of pulse, blood pressure, and electrocardiograms (ECGs) for baseline and complete treatments on consecutive days. After fasting overnight, subjects were dosed with oral phenylephrine HCl 10, 20, or 30 mg or placebo. Pharmacokinetic blood samples were collected over 7 h, whereas pulse, blood pressure, and ECGs were measured over 12 h. Urine was collected over each 24-h period to quantify phenylephrine and metabolites.
Results
After oral administration, phenylephrine was rapidly absorbed with median times to maximum plasma concentrations (t max) from 0.33 to 0.5 h. For phenylephrine HCl 10, 20, and 30 mg, the mean (standard deviation) maximum concentration (C max) was 1354 (954), 2959 (2122), and 4492 (1978) pg/mL, and total systemic exposure [area under the plasma concentration–time curve from time zero to infinity (AUC∞)] was 955.8 (278.5), 2346 (983.8), and 3900 (1764) pg·h/mL, respectively. Both parameters increased disproportionally with increasing dose, as β >1 in the power model. Negligible amounts of phenylephrine and phenylephrine glucuronide were excreted in urine. With increasing dose, percentages by dose of phenylephrine sulfate decreased, whereas percentages of 3-hydroxymandelic acid increased. Eight subjects reported nine mild adverse events; one (somnolence) was deemed to be treatment related. Means of time-matched differences in pulse and blood pressure from baseline showed similar fluctuations over 12 h among phenylephrine HCl doses and placebo, although small differences in systolic pressure were observed during the initial 2 h. No apparent dose-related effects were observed for Fridericia-corrected QT interval (QTcF) values, and individual changes from time-matched baseline (DQTcF).
Conclusions
Maximum and total systemic exposures following singe doses of phenylephrine HCl 10, 20, and 30 mg increased disproportionally with increasing dose. Safety and cardiovascular tolerability were comparable among doses and placebo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Phenylephrine HCl 10 mg, a nasal decongestant, was developed in the 1960s and not subject to current clinical and regulatory standards for market approval. Therefore, pharmacokinetic and metabolic data on therapeutic and higher doses are limited or non-existent. |
Maximum and total systemic exposures following the three doses of phenylephrine HCl (10, 20, and 30 mg) increased disproportionally with increasing dose. The increases in plasma concentrations, along with decreases in urinary excretion of the sulfate conjugate, suggest that more phenylephrine may bypass intestinal-wall metabolism at the higher doses. |
Cardiovascular tolerability, as measured by serial pulse, blood pressure, and electrocardiograms over 12 h, was comparable among single oral doses of phenylephrine HCl 10, 20, and 30 mg and placebo. |
1 Introduction
Phenylephrine HCl is indicated for the temporary relief of nasal congestion caused by the common cold or allergic rhinitis. As a vasoconstrictor, phenylephrine possesses both indirect and direct sympathomimetic effects. The dominant, direct effect is agonism at α1-adrenergic receptors located on capacitance blood vessels of the nasal mucosa, resulting in vasoconstriction, which limits the amount of fluid to enter the nose, throat, and sinus linings, and decreases inflammation of nasal membranes [1, 2]. In the USA and Australia, the adult dosage of phenylephrine HCl is 10 mg every 4 h, whereas in a few South American countries, the adult dosage in combination products may be 20 mg every 8 h. The total daily dose is 60 mg for both regions. In Europe and Russia, the adult dosage of phenylephrine HCl is 12 mg every 6 h, not to exceed 48 mg in 24 h.
The therapeutic dose of phenylephrine HCl 10 mg has no substantial stimulant effect on the β-adrenergic receptors of the heart (β1-adrenergic receptors), nor does it stimulate the β-adrenergic receptors of the bronchi or peripheral blood vessels (β2-adrenergic receptors) [1, 3]. Although oral phenylephrine has a minimal direct effect on heart rate or cardiac output, as a vasoconstrictor, phenylephrine can increase systolic and diastolic blood pressure at high doses, and thus cause reflex bradycardia [3]. A published review of single-dose studies in normotensive subjects with and without nasal congestion found that phenylephrine HCl 50 mg appears to be the minimum oral dose that will affect blood pressure, whereas 120 mg is needed to elicit significant blood pressure elevation [3]. Keys and Violante [4] reported that single oral doses of phenylephrine HCl 40–60 mg are necessary for consistent clinically meaningful cardiovascular effects, such as increased diastolic pressure and reflex cardiac slowing.
Although phenylephrine HCl 10 mg has been used worldwide for over 50 years in decongestant medicines, published pharmacokinetic and metabolic data on this therapeutic dose, and other doses, are limited or non-existent. One early study evaluated the pharmacokinetics of radiolabeled phenylephrine in a few volunteers after intravenous and oral administration [5]. It showed that phenylephrine is rapidly absorbed between 45 and 75 min, and rapidly distributes into peripheral tissues, yielding a biphasic, or two-compartment, pharmacokinetic profile. The central volume of distribution (340 ± 174 L) estimated from intravenous data considerably exceeds body weight, indicating storage in the peripheral compartment. Additionally, phenylephrine was found to have a short elimination half-life (t ½β) of about 2.5 h and a relatively low oral bioavailability of about 38 % due to high first-pass metabolism in the intestinal wall.
A subsequent pilot study (n = 12) in which phenylephrine was assayed directly in plasma samples using liquid chromatography with mass spectrometry found that maximum concentrations (C max) were reached sooner, within an average of 36 min, and t ½β was shorter, about 0.5–2.2 h, than previously reported [6]. Additionally, inter-subject variability of phenylephrine concentrations was high, as individual C max values ranged from 800 to 3400 pg/mL.
A published report on the pharmacokinetic drug interaction between phenylephrine and acetaminophen summarized and modelled data from four crossover studies, three of which included the phenylephrine HCl 10 mg dose as a single-ingredient treatment [7]. Mean C max values ranged from 873.8 to 1132 pg/mL, and mean extrapolated area under the plasma concentration–time curve from time zero to infinity (AUC∞) values ranged from 1105 to 1916 pg·h/mL.
The objectives of this study were to characterize the single-dose pharmacokinetics and safety of 10, 20, and 30 mg doses of phenylephrine HCl in healthy adults and to assess the cardiovascular tolerability of these doses compared with time-matched baseline and placebo. In addition, the metabolism of phenylephrine was evaluated using urinary excretion data.
2 Methods
2.1 Study Design
The study was conducted at Biovail Contract Research (Toronto, ON, Canada) in September 2009. It had a randomized, double-blind, placebo-controlled, single-dose, crossover design, comparing four treatments: placebo and phenylephrine HCl 10, 20, and 30 mg (Fig. 1). For blinding, placebo tablets were manufactured by McNeil Consumer Healthcare (Fort Washington, PA, USA) to visually match the 10 mg commercial Sudafed PE® tablet (Pfizer Inc., New York, NY, USA). Each treatment consisted of three tablets with the appropriate number of placebo and/or phenylephrine HCl 10 mg tablets to yield the assigned dose.

Study design scheme where phenylephrine HCl 10, 20, and 30 mg doses are designated as A, B, and C, respectively, and the placebo dose is designated as D. BP blood pressure, ECG electrocardiogram, PK pharmacokinetic
Twenty-eight healthy subjects were admitted to the clinic 2 days (Day −2) before the first treatment dose and remained housed until discharge on Day 5. Starting on the morning of Day −1, pulse, blood pressure, and 12-lead electrocardiogram (ECG) were measured intermittently over 12 h at the same times scheduled for measurements on subsequent treatment days. These values served as time-matched baselines for the respective measures on Days 1–4.
Seven subjects were randomized to each of four treatment sequences. On the morning of Days 1–4, the pharmacist dispensed the assigned treatment for each subject into a dosing container marked with the randomization number to maintain the blind. After fasting overnight, subjects swallowed their assigned treatment with 240 mL of water. Pharmacokinetic blood samples were collected over 7 h, whereas pulse, blood pressure, and 12-lead ECG were measured over 12 h. Urine was collected over each 24-h treatment period to quantify phenylephrine and metabolites. Standardized meals and beverages were free of apples, grapefruit, red grapes, spinach, broccoli, onions, and caffeine. On Day 5, a physical examination and 12-lead ECG were completed, and blood pressure and pulse were measured before subjects were discharged from the study.
2.2 Subjects
Men and women, ages 18–45 years, with a body mass index (BMI) of approximately 18–28 kg/m2 and a body weight >60 kg, were eligible to participate if they were deemed healthy by medical history, clinical laboratory tests, 12-lead ECG, and physical examination. Eligible subjects had a resting pulse of 50–100 beats per min (bpm) and were normotensive (sitting blood pressure within 90–140 mmHg systolic and 50–90 mmHg diastolic).
Prospective subjects were excluded if they had a positive test for HIV antibodies, hepatitis B surface antigen, or hepatitis C antibody, or a positive urine test for drugs of abuse. Female subjects who were pregnant or nursing, or unwilling to use an acceptable form of contraception were excluded. Other exclusion criteria included a corrected QT (QTc) interval of >450 ms for men and >470 ms for women on 12-lead ECG; history of long QT syndrome or arrhythmia; allergy or sensitivity to phenylephrine HCl; heavy alcohol consumption; history of smoking or use of nicotine-containing substances in the previous 2 months; use of food or beverages containing grapefruit, Seville oranges, or quinine within 24 h of dosing; use of any methylxanthine-containing products within 48 h of dosing; excessive use of caffeine from screening to clinic admission; use of any monoamine oxidase inhibitor within a month of dosing; and use of prescription or non-prescription medication (except for acetaminophen), vitamins, or herbal supplements within 14 days or 5 half-lives (whichever was longer) of dosing.
2.3 Sample Collection and Assays
For the pharmacokinetic analysis, blood samples were collected at 0 (predose), 10, 15, 20, 25, 30, and 40 min and 1, 1.5, 2, 3, 4, 5, 6, and 7 h after each dose. The 4 mL blood samples were drawn by venous puncture or indwelling catheter into tubes containing the anticoagulant, tripotassium ethylenediamine tetra-acetic acid, and centrifuged (3000 rpm at 4 °C) for 10 min. Supernatant plasma was divided into two aliquots and stored in polypropylene cryovials at −70 °C until assayed.
For the metabolite analysis, a 25 mL predose aliquot of urine was collected within 15 min of dosing on Day 1 after subjects completely voided their bladders. On Days 1 through 4, all urine was completely collected for 24 h, beginning immediately after the dose and ending with the final, scheduled void before the next dose. For each 24-h interval, urine was pooled into a covered container and stored refrigerated. The total amount of urine collected was measured by weight. After thorough mixing, two 25 mL aliquots were withdrawn from the pooled urine for each treatment day and stored frozen at −70 °C until assayed.
Bioanalytical assays were conducted by PPD, Inc. (Madison, WI, USA). Plasma samples were quantified for concentrations of phenylephrine (unconjugated, free base) using a validated liquid chromatography–tandem mass spectrometry (LC/MS/MS) method with quantification limits from 10.0 to 2500 pg/mL [8]. The inter-assay variability was ≤4.15 %, and the accuracy ranged from −1.20 to 2.64 % across quality control samples.
Urine samples were quantified for concentrations of phenylephrine (unconjugated, free base), total phenylephrine (unconjugated plus enzyme-digested conjugates), and two metabolites using validated LC/MS/MS assay methods [9–11]. The quantification limits were 2.0–200 ng/mL for phenylephrine, 100–5000 ng/mL for total phenylephrine, 2.0–200 ng/mL for phenylephrine glucuronide, and 100–5000 ng/mL for 3-hydroxymandelic acid. The inter-assay variability was ≤5.58, 5.78, 4.30, and 8.85 %, respectively. The accuracy ranged from 0.382 to 1.66 %, −7.41 to 4.37 %, −1.81 to −0.594 %, and −0.405 to 4.43 %, respectively.
2.4 Pharmacokinetic Analysis
Phenylephrine pharmacokinetic parameters were derived or observed from individual plasma concentrations, and they include area under the plasma concentration–time curve (AUC) to the last measurable concentration (AUClast); AUC from time zero to infinity (AUC∞); C max; time to C max (t max); terminal beta t ½β; apparent total oral clearance (CL/F); and apparent volume of distribution (V d/F). The data were analyzed with actual sampling times using WinNonlin®, version 5.2 (Pharsight Corporation, Cary, NC, USA) and non-compartmental methods (e.g., linear-log trapezoidal method for AUC and log-linear regression for t ½β). In the analysis, plasma concentrations below the limit of quantification occurring before C max were replaced by zero, and those occurring in the terminal phase was treated as missing in the dataset.
For the urine pharmacokinetic analysis, the amounts of phenylephrine and three metabolites excreted into urine over 24 h were estimated. Amounts of phenylephrine, phenylephrine glucuronide, and 3-hydroxymandelic acid were estimated directly by multiplying assayed concentrations by urine volumes collected over 24 h. Because phenylephrine sulfate could not be synthesized and assayed directly in urine, the amount excreted was estimated indirectly. An aliquot of urine was digested with sulfatase, and the concentration of total phenylephrine (unconjugated phenylephrine + phenylephrine from digestion of phenylephrine sulfate) was assayed. After multiplying the concentration by the 24-h urine volume and converting analytes to micromoles (µmol), the amount of phenylephrine sulfate was estimated by subtracting the amounts of unconjugated phenylephrine and phenylephrine glucuronide from total phenylephrine in the digested sample. The excreted amounts (Ax) were expressed as percentages of the administered phenylephrine dose (PDx), using the following equation (Eq. 1):
The free-base doses for phenylephrine HCl 10, 20, and 30 mg are 8.21 (49), 16.42 (98.0), and 24.63 mg (147 µmol), respectively.
2.5 Cardiovascular Assessments
Cardiovascular assessments included measurements of pulse, blood pressure, and ECGs over 12 h after each treatment dose. Because of diurnal variation in cardiovascular function [12–14], baseline measurements were collected on Day −1 to match scheduled timepoints on the subsequent dosing days. On Days −1 through 4, sitting pulse and blood pressure were measured at 30 min before the scheduled dosing time, and at 15, 20, 30, 40, 55, and 75 min, and 1.5, 2, 3, 4, 5, 6, 7, 8, 10, and 12 h after the dosing time. They were measured with an automated device, consisting of an inflatable cuff, oscillatory detection system, and automatic recorder. Blood pressure was measured in triplicate and the average value reported for each timepoint; whereas the initial, single measurement for pulse was reported.
On Days −1 through 4, a single ECG was recorded at 30 min before the scheduled dosing time, and at 50, 75, and 105 min, and 2.5, 3.5, 4.5, 7.5, 10, and 12 h after the dosing time. The 12-lead ECG recordings were machine-read,Footnote 1 providing values for heart rate, PR interval, QRS interval, and QT interval. Fredericia’s correction (QTcF) was applied to the QT interval data for the cardiovascular assessments, although Bazett’s correction (QTcB) was used in safety monitoring during the study [15, 16]. The recordings and output were reviewed for clinical significance, and repeated when requested by the investigator.
2.6 Safety Assessments
Subjects were screened for health status by physical examination, vital signs, ECGs, and clinical laboratory tests (hematology, chemistry, and urinalysis). Safety was assessed by observed or spontaneously reported treatment-emergent adverse events throughout the study, review of the cardiovascular measurements, and results of the physical examination with vital signs and an ECG completed at the end of the study.
2.7 Statistical Methods
Statistical analyses were conducted using SAS®, version 9.1. (SAS Institute Inc., Cary, NC, USA). The pharmacokinetic and cardiovascular safety results were summarized with descriptive statistics (e.g., arithmetic mean and standard deviation).
2.7.1 Dose Proportionality
The power model, y = α(dose)β, was used to describe the relationship between dose and a pharmacokinetic parameter (y), where a strictly dose-proportional relationship exists if β = 1. Estimates of β with 90 % confidence intervals were obtained from fitting a linear regression to log-transformed AUC∞ and C max data with log dose as a covariate (Eq. 2):
2.7.2 Pulse and Blood Pressure
Incidence counts and percentages of subjects with pulse <50 or >100 bpm, diastolic blood pressure <50 or >90 mmHg, and systolic blood pressure <90 or >140 mmHg were tabulated for baseline and each treatment, and at study end. Changes in pulse and blood pressure from time-matched baselines for 12 h following dose administration were calculated. Baseline values for each subject and time point on Day −1 were subtracted from the subject’s corresponding time-matched values on Days 1–4. These values were summarized statistically by treatment and timepoint.
2.7.3 Electrocardiograms
Incidence counts and percentages of subjects with QTcF >450 to ≤480, >480 to <500, and ≥500 ms were tabulated for baseline and each treatment, and at study end. Additionally, incidence counts and percentages of subjects with a change in QTcF from time-matched baseline (DQTcF) on Day –1 of >30 to ≤60 and >60 ms were tabulated. QTcF values for each subject and timepoint over 12 h on Day –1 were subtracted from the subject’s corresponding time-matched values on Days 1–4 to obtain the DQTcF values. However, some of the times were not closely matched between Day −1 and Days 1–4, mainly at −30 min, 50 min, and 1.25 h. Hence, the QTcF value on Day −1 that was measured closest in time to values on Days 1–4 was used as the time-matched baseline. If two values were equidistant, then the earlier one was used. DQTcF was summarized statistically by timepoint and treatment.
2.7.4 Phenylephrine Concentrations and Adjusted Fridericia-Corrected QT Interval (QTcF)
The potential for drug concentration effects on temporal changes in QTcF was explored graphically during the first 4.5 h after dosing when concentrations were highest. Phenylephrine plasma concentrations were interpolated for timepoints of QTcF values without coincident measured concentrations. Using the nonlinear trapezoidal rule in WinNonlin® (version 5.2), partial AUCs of phenylephrine were estimated between ECG measurement timepoints. The plasma concentration at each ECG timepoint was then extracted from output generated from the analysis. Next, DQTcF for each subject, treatment, and timepoint up to 4.5 h were calculated as previously described. Finally, differences in DQTcF from time-matched placebo DQTcF (DDQTcF) were calculated for each subject, timepoint, and phenylephrine dose. The DDQTcF values were plotted against the logarithm of corresponding measured and interpolated phenylephrine plasma concentrations. Note that the definition of DDQTcF is the difference in QTcF values between each phenylephrine HCl dose and placebo because the baseline values are the same for each treatment in the crossover design.
3 Results
3.1 Subjects
Twenty-eight subjects (13 male and 15 female) enrolled in the study, of whom 27 subjects completed four treatments. One subject withdrew for personal reasons after completing two treatments and partially completing the third. Available pharmacokinetic, safety, and cardiovascular data for all 28 subjects were included in the analyses. Mean (±standard deviation) age, weight, height, and BMI for the study population were 32.0 ± 6.6 years, 70.0 ± 10.5 kg, 168.5 ± 10.1 cm, and 24.6 ± 2.5 kg/m2, respectively. Racial and ethnic composition included four Asian, four black, 11 Hispanic, and nine white subjects.
3.2 Pharmacokinetics
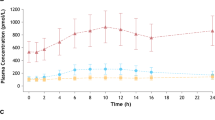
Mean plasma concentration–time profiles following single-dose administration of phenylephrine HCl 10, 20, and 30 mg are shown in Fig. 2. Phenylephrine was rapidly absorbed with the median t max ranging from 0.33 to 0.5 h across doses, after which it rapidly declined with mean terminal t ½β from 1.64 to 1.93 h (Table 1). Both C max and AUC∞ increased disproportionally with increasing doses, where estimates of β [associated 90 % confidence intervals] from the power model were 1.17 [1.02, 1.32] and 1.25 [1.15, 1.34], respectively. Consistent with the disproportionate increase in AUC∞ with dose, oral phenylephrine CL/F and V d/F decreased modestly. Estimates of t max and terminal t ½β were similar across doses.

Mean phenylephrine plasma concentration–time profiles by dose from 0 to 7 h (a), and an expanded view from 0 to 4 h (b). The error bars represent standard deviations at each sampling time
In urine, the percentages of dose, PDx, excreted as unchanged phenylephrine and phenylephrine glucuronide over 24 h were negligible for the three doses of phenylephrine HCl (Table 1). PDx excreted as phenylephrine sulfate decreased from 46.6 to 36.2 % with increasing dose, whereas PDx as 3-hydroxymandelic acid increased from 25.3 to 30.2 %.
3.3 Pulse and Blood Pressure Assessments
The incidence count and percentage of subjects with pulse or blood pressure values below and above the reference limits were comparable at baseline, and after placebo and the three phenylephrine HCl doses (Table 2). Incidences were few, ranging from 0 to 12 out of approximately 475 total measurements per 28 subjects for baseline or each treatment period.
Mean changes in pulse and blood pressure from time-matched baselines following dose administration are displayed by treatment in Figs. 3 and 4, respectively. The fluctuating patterns over 12 h were similar among the three phenylephrine HCl doses and placebo. Although the mean profiles suggest no systematic dose-related effects, changes over the initial 2 h were more variable across assessments and treatments, mainly changes in systolic blood pressure from baseline for placebo in Fig 4. Later in the day, mean pulse peaked around 6 h, whereas mean blood pressure peaked at 7 h.

Mean (standard deviation) time-matched change from baseline for pulse over 12 h for the three phenylephrine HCl doses and placebo. bpm beats per min, PBO placebo

Mean (standard deviation) time-matched change from baseline for a diastolic and b systolic blood pressure over 12 h for the three phenylephrine HCl doses and placebo. BP blood pressure, PBO placebo
3.4 QTcF Assessments
The incidence counts and percentages of subjects with QTcF >450 to ≤480 ms and >480 to <500 ms were the same or lower than baseline and placebo when subjects received phenylephrine HCl 10, 20, or 30 mg (Table 3). No incidences were deemed clinically significant by the investigator. No incidents of QTcF >450 ms were observed at study end. Overall, five QTcF values >450 and <500 ms were observed when subjects received placebo. Following all phenylephrine HCl doses, five QTcF values >450 and <500 ms were observed, of which two were associated with quantifiable phenylephrine plasma concentrations. These two QTcF values (474 and 462 ms at 1.75 and 3.5 h, respectively) were observed in one female subject following the phenylephrine HCl 30 mg dose. The remaining three QTcF values were not associated with quantifiable concentrations because they were measured at 10 or 12 h after the dose (Fig. 2).
One incident of QTcF ≥500 ms was observed in the phenylephrine HCl 30 mg treatment (515 ms), although one borderline incident of 498 ms was observed in the placebo treatment. Both incidences were not deemed clinically significant. They were associated with the ECG recorded at the 10-h timepoint, when phenylephrine plasma concentrations were either negligible due to the drug’s short t ½β or non-existent, and approximately 1 h after the evening meal. Food has been shown to affect ECG parameters, including QTc prolongation, T wave shape, and heart rate, up to 1 and 4 h after consumption [17–19].
The incident counts and percentages of subjects with DQTcF >30 to ≤60 ms were similar among the phenylephrine HCl 10 and 30 mg doses and placebo, but were higher for the 20 mg dose (Table 3). No incidents of DQTcF >60 ms were observed for the 10 mg dose or at study end, whereas three, four, and five incidences were observed for placebo, and phenylephrine HCl 20 and 30 mg, respectively. The times of these observations are apparent in the box plots of DQTcF values by timepoint and treatment in Fig. 5. The pattern and variability over 12 h were similar among the three phenylephrine HCl doses and placebo, with the highest variability at the 10-h timepoint across treatments when several of DQTcF values >60 ms were observed.

Box plots for DQTcF over 12 h for a 10, b 20, and c 30 mg doses of phenylephrine HCl and d placebo. The rectangle spans the first to third quartiles with the median segment inside, while the whiskers span 1.5 times the interquartile range. The solid circles are outliers. DQTcF difference in Fridericia-corrected QT interval from time-matched baseline
3.5 Relationship Between Phenylephrine Concentrations and Adjusted QTcF
Differences in time-matched, baseline-adjusted QTcF between phenylephrine HCl doses and placebo (DDQTcF) are plotted against corresponding measured or interpolated phenylephrine concentrations on the logarithm scale in Fig. 6. Data for the three phenylephrine HCl doses and timepoints up to 4.5 h after the dose were included. The individual data show considerable overlap, with no apparent trend in DDQTcF response to phenylephrine concentrations over this range.

Scatter plot of DDQTcF versus measured and interpolated phenylephrine concentrations on the logarithm scale for timepoints up to 4.5 h after the dose. DDQTcF difference in Fridericia-corrected QT interval values between each phenylephrine HCl dose and placebo
3.6 Safety
Over the range of single phenylephrine HCl doses evaluated in this study, no cardiovascular assessments outside of reference limits were deemed clinically significant by the investigator. No serious adverse events were reported, nor did any subject discontinue the study due to an adverse event. During the study, eight out of 28 subjects reported nine mild, treatment-emergent adverse events that resolved with no action. Following the placebo dose, elbow rash was experienced by one subject. Following the phenylephrine 10 mg dose, one instance each of dizziness, headache, and arm pain was experienced by three subjects. Following the phenylephrine 20 mg dose, one instance each of somnolence, arm bruise, and elbow rash was experienced by three subjects. Following the phenylephrine 30 mg dose, one instance each of fatigue and dizziness was experienced by two subjects. One adverse event, somnolence, was deemed possibly related to treatment by the investigator, and the other eight adverse events were deemed unlikely related or unrelated.
4 Discussion
Following single-dose oral administration, phenylephrine was rapidly absorbed from the 10, 20, and 30 mg doses, with C max occurring between 13 and 60 min. This was followed by a rapid decline in plasma concentrations due to phenylephrine’s short t ½β, resulting in minimal concentrations beyond 4 h, when phenylephrine HCl would typically be re-dosed. Similar to previous studies of the 10 mg dose, inter-subject variability for maximum and total exposures of phenylephrine (C max and AUC∞) was high [6, 7]. At each dose, about a tenfold range in individual C max values was observed.
Both C max and AUC∞ increased more than proportionally with increasing phenylephrine HCl doses, although the disproportional increase was relatively modest. Given that phenylephrine is a drug with high first-pass metabolism, this disproportional increase in AUC∞ suggests that more phenylephrine may bypass the intestinal wall at the higher doses. The increase in bioavailability (F) with dose is consistent with the observed decreases in CL/F and V d/F with no change in the terminal t ½β.
Sulfate conjugation of phenylephrine in the intestinal wall is the main route of the first-pass metabolism following oral administration [5]. Another major route of metabolism is deamination by the A and B forms of monoamine oxidase to aldehydes, which are further metabolized to 3-hydroxymandelic acid and m-hydroxyphenylglycol [5, 20]. Conjugation of phenylephrine by glucuronide was reported as a minor metabolic pathway [20].
The percentage of phenylephrine excreted unchanged in urine was very low at <0.5 % for the three phenylephrine HCl doses. Unlike a previous study reporting about 6 % phenylephrine glucuronide, this metabolite was excreted at <0.1 % for all doses [20]. Although m-hydroxyphenylglycol and phenylephrine sulfate were not measured in this study, amounts of the sulfate conjugate excreted were estimated indirectly after sulfatase digestion of urine samples. The percentages of phenylephrine sulfate and 3-hydroxymandelic acid across doses were similar to the respective 47 and 30 % reported previously following oral administration [5]. With increasing phenylephrine HCl doses, percentages of the sulfate conjugate excreted decreased, whereas percentages of 3-hydroxymandelic acid increased. This decrease in pre-systemic sulfation may partly explain the greater than proportional increases in phenylephrine plasma concentrations with increasing doses.
The categorical assessments of pulse and blood pressure outside reference limits were comparable among the single oral doses of phenylephrine HCl 10, 20, and 30 mg, placebo, and baseline. Means of time-matched differences in pulse and blood pressure from baseline showed similar fluctuations over 12 h among phenylephrine HCl doses and placebo. Initially, small increases in mean blood pressure up to 40 min coincided with small decreases in mean pulse up to 20 min. Maximum increases in mean pulse and blood pressure were observed between 6 and 7 h, respectively, for all treatments, reflecting the acrophase or high point of diurnal variation that typically occurs daily at around 16:00 h [13].
The main purpose of serial measurements of pulse, blood pressure, and ECGs was to assess cardiovascular tolerability of single doses of phenylephrine at the main therapeutic and two higher doses. As such, this study was not designed to thoroughly evaluate the QTc interval, which would require multiple dosing, inclusion of a positive control, replicate ECGs at each timepoint to lower variability, and ECG over-read by qualified cardiologists [14, 16]. Therefore, these results do not appropriately address potential proarrhythmic risk due to phenylephrine.
In a meta-analysis of ECG data from baseline and placebo treatments in phase I studies, the incidence and percent of male subjects with absolute QTcF values >450 ms and increases of 30–60 ms or >60 ms from baseline were shown to increase with more occasions of ECG recordings per subject and greater variability in QTcF values [14]. In the current study, single ECGs were recorded at ten timepoints per subject during baseline and again on each subsequent treatment day. Standard deviations of QTcF means for 28 subjects across timepoints and treatments were relatively high, ranging from ±13.8 to ±28.9 ms. Therefore, in addition to the incidences of QTcF values within and above the reference limits being similar among treatments, the percentages of subjects with at least one observation within and above the categorical limits are consistent with the study design and QTcF variability.
Another consideration is that the QTcF categorical analysis included incidence counts of QTcF >450 ms as a conservative lower limit for the cardiovascular assessment. Female subjects were allowed to enroll in this study with a QTc interval ≤470 ms, which is often used as an upper reference limit for women because women have a longer QTc interval than men [21]. Twelve of the 13 QTcF values >450 ms were recorded in women and, of these, ten values were ≤470 ms. The remaining values were borderline (474 ms) and high (498 ms) in two female subjects and high (515 ms) in a male subject. Both high values were recorded 10 h after the dose when phenylephrine plasma concentrations were either negligible or non-existent, and within an hour of the evening meal [14, 18].
Overall, the cardiovascular assessments showed no, or minimal, effects following single doses of phenylephrine HCl 10, 20, and 30 mg. A review of heart rate and blood pressure from a series of placebo-controlled, nasal decongestant studies showed statistically significant differences from baseline following doses of phenylephrine HCl 10 and 25 mg [22]. However, these changes were relatively small and inconsistent with regard to increases or decreases over the measurement interval, and did not account for diurnal variation with time-matched baselines. In a study on a pharmacokinetic drug interaction between phenylephrine and acetaminophen, researchers were unable to show any change in mean arterial blood pressure in the healthy volunteers for phenylephrine concentrations in the range expected for a 20 mg dose [7]. Finally, the cardiovascular assessments in this and other studies of therapeutic doses are in accordance with published reports that found higher single oral doses from 40 to 120 mg are necessary for consistent, clinically meaningful cardiovascular effects [3, 4].
5 Conclusion
The single-dose pharmacokinetics of phenylephrine HCl 10, 20, and 30 mg in this study confirmed the rapid absorption and elimination of phenylephrine reported previously for the 10 mg dose. Maximum and total systemic exposures following the three doses of phenylephrine HCl increased disproportionally with increasing dose. The increases in plasma concentrations, along with decreases in urinary excretion of the sulfate conjugate, suggest that more phenylephrine may bypass intestinal-wall metabolism at the higher doses. Increases in oral bioavailability resulted in the observed decreases in oral CL/F and V d/F.
Single oral doses of phenylephrine HCl 10, 20, and 30 mg were well-tolerated in this study. No serious adverse events were reported, and only one event, mild somnolence, was rated as possibly related to phenylephrine HCl. No incidences of pulse, blood pressure, or QTcF outside of reference limits were deemed clinically significant. In light of the study’s design limitations and conservative limit for the QTcF categorical assessment, these results show that cardiovascular tolerability of the three phenylephrine HCl doses, which provide a wide range of phenylephrine plasma concentrations, was comparable.
Notes
Welch Allyn® CardioPerfect® Pro Recorder or the HP M1770A/Page Writer 300 PI.
References
Johnson DA, Hrick JG. The pharmacology of a-adrenergic decongestants. Pharmacotherapy. 1993;13:110S–5S.
Empey DW, Medder KT. Nasal decongestants. Drugs. 1981;21:438–43.
Chua SS, Benriomj SI. Non-prescription sympathomimetic agents and hypertension. Med Toxicol. 1988;3:387–417.
Keys A, Violante A. The cardio-circulatory effects in man of neo-synephrin. J Clin Invest. 1942;21:1–12.
Hengstmann JH, Goronzy J. Pharmacokinetics of 3H-phenylephrine in man. Eur J Clin Pharmacol. 1982;21:335–41.
Ptacek P, Macek JK. Development and validation of a liquid chromatography-tandem mass spectrometry method for the determination of phenylephrine in human plasma and its application to a pharmacokinetic study. J Chromatogr B. 2007;858:263–8.
Atkinson HC, Stanescu I, Salem II, Potts AL, Anderson BJ. Increased bioavailability of phenylephrine by co-administration of acetaminophen: results of four open-label, crossover pharmacokinetic trials in healthy volunteers. Eur J Clin Pharmacol. 2015;71:151–8.
Method P898. Quantitation of unconjugated phenylephrine in human plasma via HPLC with MS/MS detection, Issued 2009. Middleton: PPD Bioanalytical Laboratory; 2009.
Method P956. Quantitation of phenylephrine and phenylephrine glucuronide in human urine via HPLC with MS/MS detection. Issued 2009. Middleton: PPD Bioanalytical Laboratory; 2009.
Method P957. Quantitation of 3-hydroxymandelic acid in human urine via HPLC with MS/MS detection. Issued 2009. Middleton: PPD Bioanalytical Laboratory; 2009.
Method P958. Quantitation of total phenylephrine in human urine via HPLC with MS/MS detection. Issued 2009. Middleton: PPD Bioanalytical Laboratory; 2009.
Dominguez-Rodriguez A, Abreu-Gonzalez P, Kaskic J. Disruption of normal circadian rhythms and cardiovascular events. Heart Metab. 2009;44:11–5.
Vandewalle G, Middleton B, Rajaratnam S, et al. Robust circadian rhythm in heart rate and its variability: influence of exogenous melatonin and photoperiod. J Sleep Res. 2007;16:148–55.
Harris RI, Steare SE. A meta-analysis of ECG data from healthy male volunteers: diurnal and intra-subject variability, and implications for planning ECG assessments and statistical analysis in clinical pharmacology studies. Eur J Clin Pharmacol. 2006;62:893–903.
Sagie A, Larson MG, Goldberg RJ, Bengston JR, Levy D. An improved method for adjusting the QT interval for heart rate (the Framingham Heart Study). Am J Cardiol. 1992;70:797–801.
Guidance for Industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Rockville: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, ICH; 2005.
Nagy D, DeMeersman R, Gallagher D, et al. QTc interval (cardiac repolarization): lengthening after meals. Obesity Res. 1997;5:531–7.
Widerlov E, Jostell KG, Claesson L, et al. Influence of food intake on electrocardiograms of healthy male volunteers. Eur J Clin Pharmacol. 1999;55:619–24.
Pharmaceutical Research and Manufacturers of America QT Statistics Expert Working Team. Investigating drug-induced QT and QTc prolongation in the clinic: a review of statistical design and analysis considerations: report from the pharmaceutical research and manufacturers of America QT statistics expert team. Drug Inf J. 2005;39:243–66.
Ibrahim KE, Midgley JM, Crowley JR, Williams CM. The mammalian metabolism of R-(-)-m-synephrine. J Pharm Pharmacol. 1983;35:144–7.
Rautaharju PM, Zhang ZM, Haisty WK, et al. Race- and sex-associated differences in rate-adjusted QT, QTpeak, ST elevation and other regional measures of repolarization: the Atherosclerosis Risk in Communities (ARIC) study. J Electrocardiol. 2014;47:342–50.
Consumer Healthcare Products Association. Briefing book: meeting of the Nonprescription Drugs Advisory Committee, December 2007 [Docket no. 2007P-0047]. p. 40–45. http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4335b1-03-CHPA.pdf. Accessed 21 Apr 2015.
Acknowledgments
The authors acknowledge contributions of former employees: Dolly Parasrampuria, PhD, for study design and oversight; Katherine McGinley, BS, for clinical operations; and Hiral Mankad, PharmD, for manuscript development.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored and funded by McNeil Consumer Healthcare, a Division of Johnson & Johnson Consumer, Inc. Editorial support was provided by Kathleen Boyle, PhD, from KE Boyle Consultants, LLC (Exton, PA, USA), and funded by McNeil Consumer Healthcare.
Conflict of interest
Cathy Gelotte and Brenda Zimmerman were employees at the time the study was conducted, and hold stock or stock options in Johnson & Johnson.
Ethical approval
The study was conducted in accordance with guidelines of the International Conference on Harmonisation–Good Clinical Practice and local Canadian laws on clinical research involving human subjects. The protocol and informed consent form were reviewed and approved by the Ontario Institutional Review Board (Aurora, ON, Canada).
Informed consent
All subjects were informed of the nature and purpose of the study, and gave written informed consent to participate before any screening procedures.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gelotte, C.K., Zimmerman, B.A. Pharmacokinetics, Safety, and Cardiovascular Tolerability of Phenylephrine HCl 10, 20, and 30 mg After a Single Oral Administration in Healthy Volunteers. Clin Drug Investig 35, 547–558 (2015). https://doi.org/10.1007/s40261-015-0311-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-015-0311-9