
Abstract
The voluntary withdrawal of Vioxx (rofecoxib) from the market in 2004, as well as the 2005 and 2014 US FDA Advisory Committee meetings about non-steroidal anti-inflammatory drugs (NSAIDs) and cardiovascular risk, have raised questions surrounding the use of NSAIDs in at-risk populations. This paper discusses the cardiovascular safety profile of naproxen in the context of the NSAID class. The balance of evidence suggests that cardiovascular risk correlates with cyclooxygenase (COX)-2 selectivity, and the low COX-2 selectivity of naproxen results in a lower cardiovascular risk than that of other NSAIDs. The over-the-counter (OTC) use of naproxen is expected to pose minimal cardiovascular risk; however, the benefit–risk ratio and appropriate use should be considered at an individual patient level, particularly to assess underlying conditions that may increase the risk of events. Likewise, regulatory authorities should revisit label information periodically to ensure labeling reflects the current understanding of benefits and risks.
Similar content being viewed by others

The totality of evidence suggests that while non-steroidal anti-inflammatory drugs (NSAIDs) likely increase the risk of cardiovascular events, they do so based on cyclooxygenase (COX)-2 selectivity, with greater affinity for COX-2 imparting greater risk. |
Naproxen has low COX-2 selectivity, instead demonstrating greater selectivity for COX-1 inhibition, imparting a consistent and demonstrably favorable thromboembolic and overall cardiovascular safety profile among the most commonly used non-aspirin NSAIDs. |
1 Introduction
1.1 Non-Steroidal Anti-Inflammatory Drug (NSAID) Background
1.1.1 Therapeutic Importance
Musculoskeletal aches and pains are one of the most common medical complaints around the world, and increasing life expectancies are driving an increased incidence of degenerative joint disease, burdening patients and healthcare systems [1]. Non-steroidal anti-inflammatory drugs (NSAIDs) are the most commonly used class of analgesic drugs, with approximately 30 million users worldwide daily [2] and over 100 million prescriptions every year in the USA [3]. NSAIDs continue to be one of the most effective and widely used forms of non-surgical pain relief for osteoarthritis [1]. A significant portion of the population appropriately manages pain with over-the-counter (OTC) NSAIDs [4]. In contrast, other prescription pain relievers (e.g., opioids) lend themselves to abuse, which has become a growing epidemic [5].
1.1.2 Regulatory Interest
The widespread use of NSAIDs means there is significant regulatory interest in this therapeutic category. Furthermore, after the voluntary withdrawal of Vioxx (rofecoxib) in 2004, regulatory authorities have focused on potential adverse cardiovascular outcomes.
The US FDA Advisory Committee meetings in both 2005 and 2014 concluded that NSAIDs increased the risk of myocardial infarction (MI) in high-risk individuals, and they supported the need for additional label warnings and studies to further clarify whether the increased risk was truly a class effect or the result of cyclooxygenase (COX)-2 selectivity. The 2005 meeting resulted in changes to the label for all NSAIDs, including OTC NSAIDs, to highlight this risk [6–8].
The 2014 FDA Advisory Committee meeting on the cardiovascular risk of NSAIDs included an FDA review of data available after 2005, highlighting a potential lower cardiovascular risk with naproxen than with other NSAIDs, as well as a discussion of the progress of PRECISION (Prospective Randomized Evaluation of Celecoxib Integrated Safety versus Ibuprofen Or Naproxen), an ongoing cardiovascular safety study [9]. The committee reaffirmed the position that class labeling was appropriate and should not differentiate between products, forms, and dose (including for OTC medications). Nonetheless, many of the committee members expressed the view that the data suggest a more favorable cardiovascular risk profile for naproxen than for other NSAIDs, even if it did not meet the evidentiary standard for supporting a regulatory label change. Furthermore, risk may be mitigated through low doses or a shorter duration of use, such as that with OTC naproxen. In addition, numerous other regulatory bodies have contributed to NSAID safety, with ingredient-specific differences in recommendations by country. For instance, in the UK, diclofenac was switched back from OTC to prescription status based on safety concerns, and the European Medicines Agency (EMA) determined that naproxen “may be associated with a lower risk for arterial thrombotic events than COX-2 inhibitors and other NSAIDs, but a small risk cannot be excluded” [10].
We review the totality of evidence regarding naproxen and cardiovascular safety in the context of NSAIDs as a class.
1.2 Naproxen Background
1.2.1 History
Naproxen has been available as a prescription product in the USA since 1976, and naproxen sodium has been approved for OTC use in many countries. Non-prescription dosing is appropriate every 8–12 h, with a maximum total daily OTC dose of 440–660 mg, as approved by local regulatory authorities. This differs from the prescription dosing regimen, which is usually 500 mg two to three times daily with a maximum total daily dose of 1500 mg.
Little difference in acute or chronic pain relief has been demonstrated between traditional NSAIDs, such as naproxen, and COX-2 selective NSAIDs (coxibs) [11, 12]. However, many of the studies comparing the efficacy of traditional NSAIDs and coxibs were inadequately powered to detect small differences between the compounds, should they exist [13]. In contrast, a recent network analysis found a significant difference between NSAIDs and acetaminophen in the treatment of pain in knee and hip osteoarthritis. The authors concluded that NSAIDs delivered clinically meaningful pain relief versus placebo but that acetaminophen has no role in the treatment of osteoarthritic pain [14].
2 Clinical Pharmacology of NSAIDs
2.1 Pharmacodynamics of NSAIDs
The major mechanism of action of NSAIDs is the blockage of prostanoid biosynthesis via inhibition of prostaglandin G/H synthase or COX [13]. COX-1 and COX-2 isoforms catalyze the initial steps of conversion of free arachidonic acid (AA) to prostaglandins with roles in nociception, hypothalamic regulation of body temperature, inflammation, hemostasis, and cardiovascular function. The most common adverse effects of NSAIDs are also largely mediated through effects on the production of prostanoids, e.g., maintaining gastrointestinal and renal homeostasis.
Despite similarities between the structure and function of COX-1 and COX-2, they each play different roles in the body [15]. The role of COX-1 is to maintain a basal rate of prostanoid biosynthesis [13], including the constitutive synthesis of prostaglandin (PG)-E2 by the gastrointestinal tract to mediate gastro-protection from stomach acid and maintain gastrointestinal homeostasis, and generation of thromboxane A2 (TXA2) by activated platelets in response to injury [12, 15]. In contrast, COX-2 activation is involved in the production of prostanoids in response to inflammatory mediators [16, 17] and in vasoprotection [12, 18].
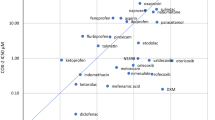
2.2 Differences in COX-2 Selectivity
NSAIDs are often categorized as non-selective or selective NSAIDs based on COX inhibition. The non-selective NSAIDs, also referred to as traditional NSAIDs (tNSAIDs), inhibit both COX-1 and COX-2 enzymes; naproxen, diclofenac, and ibuprofen are some examples of non-selective NSAIDs (Fig. 1). The relative specificity for COX-1 varies among non-selective NSAIDs. The selective COX-2 inhibitors (also referred to as coxibs), such as celecoxib, lumiracoxib, and etoricoxib, selectively inhibit the COX-2 enzyme with low degrees of COX-1 inhibition. Since COX-1 activity promotes platelet aggregation, selective COX-2 inhibitors do not have antiplatelet effects.

Non-steroidal anti-inflammatory drug cyclooxygenase selectivity and half-life. Adapted from Goodman and Gilman [19]. COX cyclooxygenase, NSAID non-steroidal anti-inflammatory drug
Naproxen binds reversibly with COX-1 and COX-2 to exert its effects but has an increased selectivity for COX-1 inhibition, which is fivefold greater than the level of COX-2 inhibition [19]. Naproxen reaches peak plasma concentrations (C max) between 2 and 4 h (naproxen sodium C max at 1–2 h) and has a half-life of 12–17 h [20]. Naproxen is a highly effective analgesic, and its long half-life provides consistent blood levels and efficacy, making it a choice comparator in many clinical trials.
The localized distribution of NSAIDs in injured tissues is necessary for maximizing therapeutic activity and lowering risks of side effects [21, 22]. NSAIDs can be categorized as acidic or non-acidic, with acidity affecting distribution of the drug. NSAIDs that are acidic (e.g., diclofenac, ibuprofen, ketoprofen) and have a high affinity for protein binding selectively accumulate at sites of inflammation [21–24], while non-acidic NSAIDs (e.g., celecoxib, rofecoxib) tend towards homogenous distribution throughout the body [21]. Naproxen, with a pKa of 4.15, falls into the acidic category [25]. These fundamental properties at least partially contribute to the effectiveness and tolerability of naproxen in the treatment of arthritis.
2.3 Implications for Cardiovascular Safety
Both COX-1 and COX-2 isozymes play an important role in the regulation of vascular homeostasis.
Platelets are an integral component of cardiovascular hemostasis and express only COX-1, in contrast to endothelial cells, which express both COX-1 and COX-2. COX-1 drives production of TXA2, which causes platelet aggregation, vasoconstriction, and an increase in vascular and cardiac remodeling. Thromboxanes increase the risk of cardiovascular events when their activity level is enhanced [26]. Thus, inhibition of COX-1 mitigates production of TXA2, potentially lowering the risk of cardiovascular events.
Differential inhibition of COX isozymes is hypothesized to be the main driver of the cardiovascular safety of NSAIDs, with greater COX-2 selectivity correlated with greater cardiovascular risk [27]. However, this correlation does not appear to be a direct correlation that can be identified by rank ordering based on COX-2 selectivity [28] (Fig. 2). This effect seems to at least partially depend upon the degree of platelet COX-1 inhibition, in combination with dosing interval and half-life [29]. Furthermore, the methodology used to determine COX isozyme selectivity influences interpretation of rank ordering and the estimation of a correlation with observed cardiovascular risk [30]. Additionally, other drug-specific effects are hypothesized to contribute, such as endothelial function and renal effects [28]. Future studies in the field will provide a better understanding of drug-specific aspects that affect cardiovascular safety other than COX selectivity.

Data from Fosbøl et al. [98]
Odds ratios and 95 % confidence intervals for cardiovascular death based on non-steroidal anti-inflammatory drug (NSAID) dose. Odds ratios for cardiovascular death (composite endpoint of death or myocardial infarction) in association with NSAID exposure
For the majority of non-aspirin tNSAIDs, the inhibition of COX-1 is transient and insufficient to inhibit platelet activation [31]. The exception is naproxen, which possesses a long half-life and, at high doses, strongly inhibits platelet COX-1 activity to prevent platelet aggregation [32, 33]. Furthermore, the ability of naproxen to inhibit thromboxane production and platelet aggregation with standard dosing may contribute to its better safety profile [34]. In contrast to aspirin’s irreversible modification of COX-1, naproxen is a reversible inhibitor of COX-1 [33].
COX-2 drives production of PGI2, which plays a cardioprotective role in the circulatory system, promotes vasodilation, and is a potent inhibitor of platelet aggregation and cell adhesion [31, 35, 36]. This activity is partially mediated via indirect antagonism towards the activity of thromboxanes, including TXA2. Therefore, inhibition of COX-2 is hypothesized to tip the natural balance between prothrombotic TXA2 and anti-inflammatory prostacyclin (PGI2), negating the cardioprotective effect of PGI2 [37]. However, a more recent investigation by Kirkby et al. [38]. did not find a role for COX-2 in prostacyclin production in the cardiovascular system, raising uncertainty about this hypothesis.
The cardiovascular system shares a major homeostasis mechanism with the kidneys: maintaining blood pressure. All NSAIDs can attenuate renal function via inhibition of COX-1 and/or COX-2 expressed in the kidneys [39]. It has been hypothesized that the observed increase in cardiovascular risk among NSAID users is due to increased blood pressure via COX-2 inhibition in the kidneys—an effect not observed with OTC doses [40]. Naproxen does not significantly increase systolic blood pressure, and this may contribute to its better safety profile [28, 41].
3 Aspirin Interaction
3.1 Non-Selective NSAIDs and Antiplatelet Effect of Aspirin
Concomitant use of low-dose aspirin for the prevention of cardiovascular events is frequent in patients taking NSAIDs for anti-inflammatory and analgesic effects [13], particularly in elderly patients with joint pain who may also be at increased cardiovascular risk. The co-administration of tNSAIDs, such as ibuprofen, but not coxibs, with low-dose aspirin has been shown to interfere with the antiplatelet effect of aspirin [42–46].
3.2 Naproxen and Aspirin
As noted, the majority of the NSAIDs, including naproxen, act as reversible competitive inhibitors of COX, and the duration of action for these non-selective NSAIDs is primarily related to their pharmacokinetic clearance [19]. In contrast, aspirin irreversibly inhibits COX-1 [47]. Naproxen is longer acting than most other tNSAIDs, with a plasma elimination half-life of 12–17 h [20].
A small study in healthy subjects found a single high dose of naproxen sodium 1000 mg reduced platelet aggregation in 60 % of cases after 24 h [48]. Furthermore, another study found that, while sequential dosing of naproxen sodium 220 mg twice daily and low-dose aspirin interfered with the irreversible inhibition of platelet COX-1 afforded by aspirin, the interaction was minimized when naproxen sodium was given 2 h after low-dose immediate-release aspirin [46]. However, based on this interaction, the impact of co-administration with aspirin still needs to be considered when seeking to understand the cardiovascular risk of naproxen. The potential for interaction between naproxen and aspirin depends on both the dose and the dosing schedule.
A recently completed study examined the impact of naproxen on serum thromboxane inhibition when added to aspirin therapy versus aspirin therapy alone (clinicaltrials.gov NCT02229461). The results of this study will provide further insights into whether OTC dosing regimens of naproxen can maintain sufficient inhibition of thromboxane B2 (TXB2) and platelet aggregation when added to an aspirin regimen at steady state.
3.3 Clinical Significance of Aspirin Interaction
3.3.1 NSAIDs Generally
Currently, the scientific literature has identified no ‘gold standard’ that defines the percent inhibition of serum thromboxane sufficient to achieve clinical benefit of platelet inhibition-derived prevention of secondary cardiovascular events. There is no conclusive demonstration in the published literature regarding the appropriate threshold for serum TXB2 inhibition that would result in clinically meaningful differences in the prevention of cardiovascular events such as stroke and MI.
Nevertheless, it is relatively understood that near-complete suppression of serum thromboxane is considered important for cardioprotection in people receiving low-dose aspirin, with observed TXB2 inhibition ranging from 95 to 99.5 % [32, 49–52].
In a real-world setting, it is difficult to infer the clinical impact of these interactions on the typical patient receiving NSAIDs because of the wide variability of these patients, including their medical background, and the lack of outcome data.
3.3.2 Naproxen Specifically
In its entirety, the data suggest that naproxen can interfere with the TXB2 inhibition provided by aspirin. However, there is no evidence that a single-day co-administration of an OTC naproxen dose and aspirin interferes with the antiplatelet effect of aspirin in a clinically meaningful way. In fact, when immediate-release aspirin is taken at least 2 h before naproxen, the TXB2 inhibition is barely impacted [46]. Nevertheless, the clinical relevance of the apparent interaction of naproxen/aspirin co-administration is yet to be established, primarily because real-life use of these drugs can stray far from the ‘ideal’ and controlled administration methods evaluated in the previously discussed trials.
4 Cardiovascular Safety
4.1 Thromboembolic Events
There are some indicators that NSAIDs may differentially increase thrombotic risk in patients commensurate with levels of cardiovascular basal risk: evaluating outcomes on the basis of the patient’s basal risk demonstrates that the predicted risk for cardiovascular events increased disproportionately more in high-risk patients than in low-risk patients, particularly with coxibs and diclofenac [53] (Fig. 3). This further supports the hypothesis that the differential increase in risk between NSAIDs correlates most strongly with COX-2 selectivity.

Annual absolute effects per 1000 of cyclooxygenase-2-selective non-steroidal anti-inflammatory drugs (NSAIDs) and traditional tNSAIDs at different baseline risks of major vascular events. For each drug category, the predicted annual absolute risks of major vascular events (±1 standard error) are shown for patients with predicted risk of 2.0 % (high risk) or 0.5 % (low risk) per annum of a major vascular event. Data from the CNT meta-analysis [68]. SE standard error
Thus, in retrospect, it should come as no surprise that an increased incidence of thrombotic events was observed with the COX-2 inhibitors celecoxib, rofecoxib, and valdecoxib in randomized placebo-controlled trials [54–56]. This observation initiated further review into the cardiovascular effects associated with NSAIDs. Importantly, the results of observational studies [29, 57, 58], a network meta-analysis [28], and two meta-analyses of data derived from randomized controlled trials (RCTs) of an NSAID versus placebo or an NSAID regimen versus another NSAID regimen have confirmed the initial observation of a cardiovascular risk with coxibs but also extended the concern to use of certain tNSAIDs [13, 59]. Additionally, a recent very large population-based case–control study found that the use of diclofenac, ibuprofen, rofecoxib, celecoxib, and meloxicam all significantly increased the risk of venous thromboembolism in patients with knee osteoarthritis, whereas the effects of naproxen use was indistinguishable from those of no NSAID use [60].
While much of the data suggest that rank ordering of the varying NSAID compounds with relation to cardiovascular risk is difficult, the balance of evidence suggests that naproxen results in a low cardiovascular risk level amongst NSAIDs, or possibly even neutral cardiovascular risk levels (i.e., equivalent to placebo). Two large prospective studies comparing coxibs and naproxen (ADAPT [Alzheimer’s Disease Anti-inflammatory Prevention Trial] and TARGET [Therapeutic Arthritis Research and Gastrointestinal Event Trial]) observed different cardiovascular risk profiles for naproxen versus coxibs, with TARGET demonstrating a numeric (but non-significant) decrease for naproxen (hazard ratio [HR] 1.46 for lumiracoxib vs. naproxen, 95 % confidence interval [CI] 0.89–2.37), and ADAPT suggestive of increased risk for naproxen (HR 1.63, 95 % CI 1.04–2.55) [61, 62]. An additional trial (SCOT [Standard Care versus Celecoxib Outcome Trial]) has yielded only topline results, but these preliminary findings are consistent with the totality of evidence. SCOT demonstrated no significant difference between celecoxib and tNSAIDs (grouped together): cardiovascular outcomes occurred in 1.8 % of the celecoxib arm and 2.2 % of the tNSAID arm (HR 1.12; p = 0.50). However, although serious adverse events occurred at a similar rate (5.2 % in the celecoxib arm vs. 5.8 % in the tNSAID arm), the celecoxib arm exhibited a significantly greater number of non-serious adverse events than the tNSAID arm (22 vs. 16.1 %; p < 0.001), and significantly more patients withdrew from the celecoxib treatment arm than from the tNSAID treatment arm (50.9 vs. 30.2 %; p < 0.0001) [63]. While the SCOT trial contributed to the vast body of knowledge on NSAIDs and cardiovascular risk, it could have been potentially more informative to have included a third placebo-controlled arm to understand baseline risk. Lastly, the APPROVe (Adenomatous Polyp Prevention on Vioxx) study compared the COX-2 selective rofecoxib with placebo and was terminated early due to a signal of increased cardiovascular adverse events [64].
Based on these clinical studies, which should carry more weight than observational studies, there is little evidence of a statistically significant increase in cardiovascular risk in either the naproxen (where applicable) or pooled tNSAID treatment groups. The only statistically significant results were reported in the TARGET naproxen sub-study and ADAPT. The TARGET sub-study demonstrated that naproxen had a lower risk of the composite cardiovascular outcome than did lumiracoxib in patients with osteoarthritis and a high baseline cardiovascular risk who are not receiving aspirin [65], while limited post hoc analyses of ADAPT identified an increased risk in a composite cardiovascular endpoint for naproxen compared with placebo.
Of course, with dozens of prospective and observational studies having been conducted, it is not unexpected that a few yield contrary results. However, these studies do not alter the balance of data, as is demonstrated in the numerous meta-analyses discussed above, especially the CNT (Coxib and traditional NSAID Trialists’) collaboration and McGettigan meta-analyses, which suggest that naproxen is not associated with an increased risk of cardiovascular thrombotic events [66–68] (Figs. 4 and 5). The more recent CNT meta-analysis observed that major vascular events are increased, by varying degrees, through the use of NSAIDs. The use of coxibs or diclofenac significantly increased major vascular events, major coronary events, and risk of vascular death. Ibuprofen use significantly increased major coronary events but did not significantly increase risk of vascular death or major vascular events. Naproxen use did not significantly increase the risk of major vascular events and did not result in an increased risk of vascular death [68].


Rate ratios and 95 % confidence intervals for myocardial infarction or coronary heart disease death due to non-steroidal anti-inflammatory drug. Data from the CNT meta-analysis [68]
The PRECISION trial will provide additional data regarding the cardiovascular risk of celecoxib compared with ibuprofen and naproxen [69, 70]. However, it is unclear (because of the limitations of PRECISION) whether it will impact the totality of the evidence reviewed in the CNT meta-analysis. In fact, the equipoise of the PRECISION trial was questioned by the FDA as a body of evidence indicates that naproxen exhibits a lower cardiovascular risk than other NSAIDs [71]. It should be noted that both PRECISION [72] and SCOT have limitations: the results of both trials will only be directly applicable to prescription dosing regimens (not OTC dosing regimens), aspirin interactions could undermine the interpretability of PRECISION results, and SCOT will be underpowered to provide insight into specific tNSAIDs. Furthermore, celecoxib exposure in the SCOT trial was lower than that in trials in which a cardiovascular risk was observed, supporting the general recommendation that a reduction in NSAID exposure results in a reduction in cardiovascular risk. This, of course, is applicable to all NSAIDs, including naproxen [29].
It is due to this totality of evidence of differential safety of naproxen with regard to cardiovascular outcomes that the American Heart Association and American College of Gastroenterology have issued the recommendations that naproxen should be the NSAID of choice for patients with high cardiovascular risk [73–76].
4.2 Hypertension
All NSAIDs, to some degree, alter vasodilation and sodium excretion by affecting prostanoid production (e.g., PGE2), which can result in hypertension, a risk factor for cerebrovascular and thromboembolic events [11, 77–79]. The COX isozymes are present in various tissues throughout the body and also affect hemostasis differently via prostanoids [80, 81]. Platelets are an integral component of cardiovascular hemostasis and express only COX-1, in contrast to endothelial cells, which express both COX-1 and COX-2. COX-1 drives production of TXA2, which causes platelet aggregation, vasoconstriction, and an increase in vascular and cardiac remodeling. COX-2 is necessary for the production of prostacyclin, which is a potent vasodilator, inhibiting platelet function and promoting renal sodium excretion [29, 73].
It has been hypothesized that an increased cardiovascular risk from NSAID use is due to an increase in blood pressure as a result of COX-2 inhibition in the kidneys and alteration of sodium and fluid retention [11, 49, 82]. The observed increase in blood pressure during long-term NSAID use is hypothesized to increase cardiovascular risk, as hypertension is a well-known risk factor for cardiovascular disease. This increased risk would essentially be associated with chronic exposure. Therefore, one would hypothesize that the risk would be directly related to increased blood pressure. However, data are limited on blood pressure changes due specifically to naproxen use and associated changes in resultant cardiac ischemic events; most of the data concern NSAIDs as a class.
An RCT [83], a large meta-analysis [84], and a systematic review of RCTs [85] all found small and occasionally significant increases in blood pressure due to prescription dose NSAID use. Given these data, it is not unexpected that some trials observed an increased risk of drug–drug interactions when prescription-strength tNSAIDs and antihypertensives were co-administered over a period of several weeks [86–88]. In contrast, short-term exposure to low doses of tNSAIDs (e.g., naproxen or ibuprofen) has not been shown to affect blood pressure or demonstrate any meaningful interaction with antihypertensive drugs, and it is not likely to increase the risk of cardiovascular events according to that mechanism [40, 88–90], whereas chronic exposure to NSAIDs in patients with hypertension does increase cardiac ischemic events [91]. This is reflected in the current class label.
Naproxen was observed to have the least risk among common NSAIDs for cardiovascular-related events and deaths, and the fact that it does not substantially increase systolic blood pressure may play a major role in its better safety profile [28, 41]. Also, the ability of naproxen to inhibit thromboxane production and platelet aggregation may lend further support to the better safety profile [34]. Thus, the data should be regarded in the context of the overwhelming body of data suggesting that naproxen has a neutral cardiovascular adverse event profile.
4.3 Congestive Heart Failure
Although the focus of NSAID safety with regard to cardiovascular risk has primarily been on increased thromboembolic risk, NSAIDs have also been implicated in fluid retention and worsening of heart failure via activity on renal function and the regulation of fluid balance [92]. Inhibition of COX-2 by NSAIDs, and the subsequent decrease in PGI2 and PGE2 in the renal cortex and juxtaglomerular cells, can result in a decrease in both renal blood flow and glomerular filtration rate [93]. There is also some evidence that NSAIDs inhibit aldosterone metabolism, with potential impacts on fluid retention, blood pressure, and cardiovascular remodeling [94]. Fluid retention is associated with worsening of heart failure in at-risk patients, especially the elderly [95].
Clinical trials are typically the most robust evidence of drug associations to outcomes. However, the two clinical studies published after 2005 reporting congestive heart failure (CHF) safety data during naproxen use provide no statistically significant confirmation of an increased risk of CHF in the naproxen treatment groups [61, 62]. Both studies were limited in their design for measuring CHF outcomes.
Since more observational studies have been conducted in the post-marketing period than clinical studies, they are a primary source for detecting safety signals on the real-world use of naproxen, despite their inherent limitations (retrospective design, selection bias, and confounding factors). Individually, these studies show no evidence of an increased CHF risk in patients exposed to naproxen at either low or high doses [95, 96].
As the number of studies primarily designed to assess the safety of naproxen with respect to the risk of development or progression of CHF is limited, the CNT meta-analysis provides insight. The authors included RCTs published after 2005 and reported that any NSAID use does increase the risk of CHF-related hospital admissions but that COX-2-selective inhibitors and ibuprofen use are associated with the highest risk, whereas naproxen was associated with a lower, albeit nonsignificant, risk in comparison [97] (Fig. 6).

Rate ratios and 95 % confidence intervals for hospitalizations for congestive heart failure due to non-steroidal anti-inflammatory drugs. Data from the CNT meta-analysis [68]
5 Conclusions
The totality of evidence suggests that while NSAIDs (both tNSAIDs and COX-2-selective NSAIDs) likely increase the risk of cardiovascular events, they do so to varying degrees. This differential increase in risk is hypothesized to correlate with COX-2 selectivity, although that correlation does not appear to be the sole determinant of cardiovascular risk. Thus, it is unsurprising that naproxen, which has low COX-2 selectivity, has been consistently observed to possess a low, or possibly even neutral, cardiovascular risk compared with other NSAIDs. Furthermore, risk may be mitigated through lower doses or shorter duration of use, such as that with OTC naproxen.
While emerging data are still to be considered for naproxen—including the SCOT and PRECISION trials—these studies are unlikely to significantly sway the totality of the evidence. Both trials examine prescription doses of NSAIDs, SCOT is underpowered to draw conclusions about individual tNSAIDs, and the interpretability of PRECISION could be undermined by aspirin interactions. New studies are unlikely to alter the benefit–risk assessment of OTC analgesics.
OTC naproxen is an appropriate pain reliever for individuals with minor aches and pains seeking self-medication. However, healthcare professionals and patients should receive proper education regarding the benefits and risks of naproxen and other NSAIDs, particularly in individuals who are, or may be, susceptible to cardiovascular side effects, to make the best treatment decision for a particular individual.
References
Towheed TE, Maxwell L, Judd MG, Catton M, Hochberg MC, Wells G. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;1:CD004257.
Singh G. Gastrointestinal complications of prescription and over-the-counter nonsteroidal anti-inflammatory drugs: a view from the ARAMIS database: arthritis, rheumatism, and aging medical information system. Am J Ther. 2000;7(2):115–21.
Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology. 2001;120(3):594–606.
Wilcox CM, Cryer B, Triadafilopoulos G. Patterns of use and public perception of over-the-counter pain relievers: focus on nonsteroidal antiinflammatory drugs. J Rheumatol. 2005;32(11):2218–24.
Substance Abuse and Mental Health Services Administration. Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings (Office of Applied Studies, NSDUH Series H-38A, HHS Publication No. SMA 10-4586Findings). Rockville, MD: Substance Abuse and Mental Health Services Administration; 2010.
US Food and Drug Administration. Joint meeting of the arthritis advisory committee and the drug safety and risk management advisory committee briefing document. Silver Spring, MD: US FDA; 2005. Available at: http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4090b1-01.htm
US Food and Drug Administration. Joint meeting of the arthritis advisory committee and the drug safety and risk management advisory committee briefing document. Silver Spring, MD: US FDA; 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM383180.pdf
FitzGerald G. Presentation to the Joint Meeting of the Joint Meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee. Mechanistic basis for a cardiovascular hazard from NSAIDs. 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM386452.pdf
Nissen S. Presentation to the Joint Meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee. The Precision Trial. 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM386446.pdf
European Medicines Agency. Assessment report for Non-steroidal anti-inflammatory drugs (NSAIDs) and cardiovascular risk. London: EMA; 2012.
FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–42.
Patrono C, Patrignani P, García Rodríguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001;108:7–13.
Bruno A, Tacconelli S, Patrignani P. Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic Clin Pharmacol Toxicol. 2014;114:56–63.
da Costa BR, Reichenbach S, Keller N, Nartey L, Wandel S, Jüni P, Trelle S. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2016;387:2093–105.
Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82:85–94.
Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Ann Rev Biochem. 2000;69:145–82.
Kean WF, Rainsford KD, Kean IR. Management of chronic musculoskeletal pain in the elderly: opinions on oral medication use. Inflammopharmacology. 2008;16:53–75.
Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s.
Goodman and Gilman (eds.). The pharmacological basis of therapeutics anti-inflammatory, antipyretic, and analgesic agents; Pharmacotherapy of Gout. 12th ed. New York: Pergamon Press; 2011.
DeArmond B, Francisco CA, Lin JS, et al. Safety profile of over-the-counter naproxen sodium. Clin Ther. 1995;17:587–601.
Brune K, Renner B, Hinz B. Using pharmacokinetic principles to optimize pain therapy. Nat Rev Rheumatol. 2010;6(10):589–98.
Brune K, Furst DE. Combining enzyme specificity and tissue selectivity of cyclooxygenase inhibitors: towards better tolerability? Rheumatology (Oxford). 2007;46(6):911–9.
Brune K. Persistence of NSAIDs at effect sites and rapid disappearance from side-effect compartments contributes to tolerability. Curr Med Res Opin. 2007;23(12):2985–95.
Rolf C, Engström B, Beauchard C, Jacobs LD, Li Liboux A. Intra-articular absorption and distribution of ketoprofen after topical plaster application and oral intake in 100 patients undergoing knee arthroscopy. Rheumatology (Oxford). 1999;38(6):564–7.
Li X, Cooper MA. Measurement of drug lipophilicity and pKa using acoustics. Anal Chem. 2012;84(6):2609–13.
Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64(4):465–74.
Maxwell SR, Payne RA, Murray GD, Webb DJ. Selectivity of NSAIDs for COX-2 and cardiovascular outcome. Br J Clin Pharmacol. 2006;62(2):243–5.
Trelle S, Reichenbach S, Wandel S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086.
Garcia-Rodriguez LA, Tacconelli S, Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J Am Coll Cardiol. 2008;52:1628–36.
Knights KM, Mangoni AA, Miners JO. Defining the COX inhibitor selectivity of NSAIDs: implications for understanding toxicity. Expert Rev Clin Pharmacol. 2010;3(6):769–76.
Patrignani P, Tacconelli S, Bruno A, Sostres C, Lanas A. Managing the adverse effects of nonsteroidal anti-inflammatory drugs. Expert Rev Clin Pharmacol. 2011;4:605–21.
Reilly IA, Fitzgerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69(1):180–6.
Capone ML, Tacconelli S, Sciulli MG, et al. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation. 2004;109(12):1468–71.
Kean WF, Lock CJ, Rischke J, Butt R, Buchanan WW, Howard-Lock H. Effect of R and S enantiomers of naproxen on aggregation and thromboxane production in human platelets. J Pharm Sci. 1989;78:324–7.
Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116(1):4–15.
Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–66.
Cheng JW. Use of non-aspirin nonsteroidal anti-inflammatory drugs and the risk of cardiovascular events. Ann Pharmacother. 2006;40:1785–96.
Kirkby NS, Lundberg MH, Harrington LS, et al. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A. 2012;109(43):17597–602. doi:10.1073/pnas.1209192109
Weir MR. Renal effects of nonselective NSAIDs and coxibs. Cleve Clin J Med. 2002;69(Suppl 1):S153–8.
Moore N, Salvo F, Duong M, Blin P, Pariente A. Cardiovascular risks associated with low-dose ibuprofen and diclofenac as used OTC. Expert Opin Drug Saf. 2014;13(2):167–79.
Sowers JR, White WB, Pitt B, et al. Rofecoxib, but not celecoxib or naproxen, increases mean 24-hour systolic blood pressure: results of a randomized double blind controlled trial in treated hypertensive patients with osteoarthritis (OA) and type 2 diabetes mellitus. Am J Hypertens. 2003;16(S1):OR-25.
Catella-Lawson F, Crofford LJ. Cyclooxygenase inhibition and thrombogenicity. Am J Med. 2001;110(Suppl 3A):S28–32.
MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573–4.
Renda G, Tacconelli S, Capone ML, et al. Celecoxib, ibuprofen, and the antiplatelet effect of aspirin in patients with osteoarthritis and ischemic heart disease. Clin Pharmacol Ther. 2006;80(3):264–74.
Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295–301.
Anzellotti P, Capone ML, Jeyam A, et al. Low-dose naproxen interferes with the antiplatelet effects of aspirin in healthy subjects: recommendations to minimize the functional consequences. Arthritis Rheum. 2011;63:850–9.
Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287–94.
Cronberg S, Wallmark E, Söderberg I. Effect on platelet aggregation of oral administration of 10 non-steroidal analgesics to humans. Scand J Haematol. 1984;33:155–9.
Patrignani P, Tacconelli S, Piazuelo E, et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost. 2014;12(8):1320–30.
Santilli F, Rocca B, De Cristofaro R, et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: implications for aspirin “resistance”. J Am Coll Cardiol. 2009;53(8):667–77.
Yokoyama H, Yaguchi T, Suzuki Y, et al. Theoretical investigation of aspirin dosage regimen to exhibit optimal antiplatelet effects and decrease risk of upper gastrointestinal lesions. Biol Pharm Bull. 2012;35(12):2112–8.
US Food and Drug Administration. Information for healthcare professionals: comcomitant use of ibuprofen and aspirin. September 2006. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM161282.pdf. Accessed Feb 2016.
Zhang J, Ding EL, Song Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials. JAMA. 2006;296:1619–32.
Bresalier RS, Sandler RS, Quan H. APPROVe Trial Investigators. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–102.
Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081–91.
Ott E, Nussmeier NA, Duke PC, et al. Efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2003;125:1481–92.
Bueno H, Bardají A, Patrignani P, Martín-Merino E, García-Rodríguez LA, Spanish Case-Control Study to Assess NSAID-Associated ACS Risk Investigators. Use of non-steroidal antiinflammatory drugs and type-specific risk of acute coronary syndrome. Am J Cardiol. 2010;105:1102–6.
Helin-Salmivaara A, Virtanen A, Vesalainen R, et al. NSAID use and the risk of hospitalization for first myocardial infarction in the general population: a nationwide case-control study from Finland. Eur Heart J. 2006;27:1657–63.
Patrono C, Baigent C. Nonsteroidal anti-inflammatory drugs and the heart. Circulation. 2014;129:907–16.
Lee T, Lu N, Felson DT, et al. Use of non-steroidal anti-inflammatory drugs correlates with the risk of venous thromboembolism in knee osteoarthritis patients: a UK population-based case-control study. Rheumatology. 2016;55(6):1099–105.
Farkouh ME, Kirshner H, Harrington RA, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet. 2004;364:675–84.
ADAPT Research Group. Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials. 2006;1(7):e33.
ESC Press Release. 2015. http://www.escardio.org/The-ESC/Press-Office/Press-releases/Last-5-years/scot-study-quells-concerns-about-nsaid-safety. Accessed Feb 2016.
Baron JA, Sandler RS, Bresalier RS, et al. Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. Lancet. 2008;372:1756–64.
Farkouh ME, Greenberg JD, Jeger RV, et al. Cardiovascular outcomes in high risk patients with osteoarthritis treated with ibuprofen, naproxen or lumiracoxib. Ann Rheum Dis. 2007;66:764–70.
McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–44.
McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8:e1001098.
Bhala N, Emberson J, Merhi A, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–79.
US National Institutes of Health. Prospective Randomized Evaluation of Celecoxib Integrated Safety versus Ibuprofen Or Naproxen (PRECISION). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT00346216. Accessed 6 Apr 2015.
Pfizer Inc. Assessment of Cardiovascular Safety in NSAIDs Advisory Committee Briefing Document. 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM386446.pdf.
Food and Drug Administration. Briefing Information for the February 10–11, 2014 Joint Meeting of the Arthritis Advisory Committee (AAC) and Drug Safety and Risk Management Advisory Committee (DSaRM). US Food and Drug Administration. 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM383180.pdf. Accessed 1 Feb 2016.
Gaffney M. Statistical issues in the design, conduct and analysis of two large safety studies. Clin Trials. 2016;13(5):513–8.
Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–42.
Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation task force on clinical expert consensus documents. J Am Coll Cardiol. 2008;52:1502–7.
Chan FK, Abraham NS, Scheiman JM, Laine L. Management of patients on nonsteroidal anti-inflammatory drugs: a clinical practice recommendation from the First international working party on gastrointestinal and cardiovascular effects of nonsteroidal anti-inflammatory drugs and anti-platelet agents. Am J Gastroenterol. 2008;103:2908–18.
Rostam A, Moayyedi P, Hunt R; Canadian Association of Gastroenterology Consensus Group. Canadian consensus guidelines on long-term nonsteroidal anti-inflammatory drug therapy and the need for gastroprotection: benefits versus risks. Aliment Pharmacol Ther. 2009;29:481–496.
Bowman TS, Gaziano JM, Kase CS, Sesso HD, Kurth T. Blood pressure measures and risk of total, ischemic, and hemorrhagic stroke in men. Neurology. 2006;67(5):820–3.
Yu Y, Stubbe J, Ibrahim S, et al. Cyclooxygenase-2-dependent prostacyclin formation and blood pressure homeostasis: targeted exchange of cyclooxygenase isoforms in mice. Circ Res. 2010;106(2):337–45.
Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276–9.
Timmers L, Sluijter JP, Verlaan CW, et al. Cyclooxygenase-2 inhibition increases mortality, enhances left ventricular remodeling, and impairs systolic function after myocardial infarction in the pig. Circulation. 2007;115:326–32.
Francois H, Athirakul K, Howell D, et al. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005;2(3):201–7.
Polonia J, Boaventura I, Gama G, et al. Influence of non-steroidal anti-inflammatory drugs on renal function and 24 h ambulatory blood pressure-reducing effects of enalapril and nifedipine gastrointestinal therapeutic system in hypertensive patients. J Hypertens. 1995;13:925–31.
Klassen D, Goodfriend TL, Schuna AA, Young DY, Peterson CA. Assessment of blood pressure during treatment with naproxen or ibuprofen in hypertensive patients treated with hydrochlorothiazide. J Clin Pharmacol. 1993;33(10):971–8.
Pope JE, Anderson JJ, Felson DT. A meta-analysis of the effects of nonsteroidal anti-inflammatory drugs on blood pressure. Arch Intern Med. 1993;153(4):477–84.
Morrison A, Ramey DR, van Adelsberg J, et al. Systematic review of trials of the effect of continued use of oral non-selective NSAIDs on blood pressure and hypertension. Curr Med Res Opin. 2007;23(10):2395–404.
Palmer R, Weiss R, Zusman RM, Haig A, Flavin S, McDonald B. Effects of nabumetone, celecoxib, and ibuprofen on blood pressure control in hypertensive patients on angiotensin converting enzyme inhibitors. Am J Hypertens. 2003;16(2):135–9.
Gurwitz JH, Everitt DE, Monane M, et al. The impact of ibuprofen on the efficacy of antihypertensive treatment with hydrochlorothiazide in elderly persons. J Gerontol A Biol Sci Med Sci. 1996;51(2):M74–9.
Houston MC, Weir M, Gray J, et al. The effects of nonsteroidal anti-inflammatory drugs on blood pressures of patients with hypertension controlled by verapamil. Arch Intern Med. 1995;155(10):1049–54.
Moore RA, Derry S, McQuay HJ. Cyclo-oxygenase-2 selective inhibitors and nonsteroidal anti-inflammatory drugs: balancing gastrointestinal and cardiovascular risk. BMC Musculoskelet Disord. 2007;8:73.
Furey SA, Vargas R, McMahon FG. Renovascular effects of nonprescription ibuprofen in elderly hypertensive patients with mild renal impairment. Pharmacotherapy. 1993;13(2):143–8.
Bavry AA, Khaliq A, Gong Y, Handberg EM, Cooper-Dehoff RM, Pepine CJ. Harmful effects of NSAIDs among patients with hypertension and coronary artery disease. Am J Med. 2011;124(7):614–20.
Aw TJ, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165:490–6.
Tegeder I, Geisslinger G. Cardiovascular risk with cyclooxygenase inhibitors: general problem with substance specific differences? Naunyn Schmiedebergs Arch Pharmacol. 2006;373(1):1–17.
Knights KM, Winner LK, Elliot DJ, Bowalgaha K, Miners JO. Aldosterone glucuronidation by human liver and kidney microsomes and recombinant UDP-glucuronosyltransferases: inhibition by NSAIDs. Br J Clin Pharmacol. 2009;68(3):402–12.
Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169(2):141–9.
Mangoni AA, Woodman RJ, Gaganis P, Gilbert AL, Knights KM. Use of non-steroidal anti-inflammatory drugs and risk of incident myocardial infarction and heart failure, and all-cause mortality in the Australian veteran community. Br J Clin Pharmacol. 2010;69:689–700.
Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–79.
Fosbøl EL, Gislason GH, Jacobsen S, Folke F, Hansen ML, Schramm TK, Sørensen R, Rasmussen JN, Andersen SS, Abildstrom SZ, Traerup J, Poulsen HE, Rasmussen S, Køber L, Torp-Pedersen C. Risk of myocardial infarction and death associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs) among healthy individuals: a nationwide cohort study. Clin Pharmacol Ther. 2009;85(2):190–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dominick J. Angiolillo has received consulting fees or honoraria from Bayer, Amgen, Sanofi, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Merck, Pfizer, and PLx Pharma; and payment as an individual from CeloNova, Johnson & Johnson, and St. Jude Medical for participation in review activities. He has also received institutional payments for grants from GlaxoSmithKline, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Janssen Pharmaceuticals, Inc., Osprey Medical, Inc., Merck, Novartis, CSL Behring, and Gilead. Steven M. Weisman is Head of Clinical and Regulatory Support at Innovative Science Solutions, a consultancy to the pharmaceutical industry, and has received consultancy fees from Bayer related to the topic of this manuscript.
Funding
No external funding was used in the preparation of this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Angiolillo, D.J., Weisman, S.M. Clinical Pharmacology and Cardiovascular Safety of Naproxen. Am J Cardiovasc Drugs 17, 97–107 (2017). https://doi.org/10.1007/s40256-016-0200-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-016-0200-5