
Abstract
Coronavirus spread is an emergency reported globally, and a specific treatment strategy for this significant health issue is not yet identified. COVID-19 is a highly contagious disease and needs to be controlled promptly as millions of deaths have been reported. Due to the absence of proficient restorative alternatives and preliminary clinical restrictions, FDA-approved medications can be a decent alternative to deal with the coronavirus malady (COVID-19). The present study aims to meet the imperative necessity of effective COVID-19 drug treatment with a computational multi-target drug repurposing approach. This study focused on screening the FDA-approved drugs derived from the fungal source and its derivatives against the SARS-CoV-2 targets. All the selected drugs showed good binding affinity towards these targets, and out of them, bromocriptine was found to be the best candidate after the screening on the COVID-19 targets. Further, bromocriptine is analyzed by molecular simulation and MM-PBSA study. These studies suggested that bromocriptine can be the best candidate for TMPRSS2, Main protease, and RdRp protein.
Similar content being viewed by others
Introduction
Coronavirus disease (COVID-19) is a public health emergency across the globe that emerged recently in Hubei province, P.R. China (Lupia et al. 2020; Al-Tawfiq 2020). The causative agent of COVID-19 is a new virus that which is thought to be emerged from SARS coronavirus (SARS-CoV), is highly contagious and pathogenic, known as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (Andersen et al. 2020). In a short period, COVID-19 has spread all over the globe. Soon it is announced as a pandemic by WHO (https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19-11-march-2020). To date, this pandemic disease has approximately 40 million confirmed cases of infection with 1.1 million confirmed deaths across the globe (https://www.worldometers.info/coronavirus/) since the first patient was hospitalized on 12 December 2019, thus stressing the menace caused by this deadly disease. Most of the infections resolve on their own without any specific treatment. Still, the immunosuppressed, and older adults with cardiovascular disease and metabolic syndrome, upon getting infected, may develop severe pneumonia, requiring immediate medical attention following treatment with supplemental oxygen (Chen et al. 2020; Zhou et al. 2020a, b).
A causative agent of various respiratory, hepatic, enteric, and neurological diseases, SARS-CoV is a positive-sense single-stranded RNA virus (Chan et al. 2013; Zumla et al. 2016). The clinical manifestation of COVID-19 ranges from asymptomatic to severe respiratory illness (Li et al. 2020a, b). The most common symptoms include dry cough, pyrexia, fatigue, and dyspnea. These symptoms may get worsen, leading to pneumonia and pleuritic chest pain that has also been reported (Chen et al. 2020). Based on symptom severity, patients can be classified into mild, severe, and critical types (Wu et al. 2020). SARS-CoV-2 has high genomic similarity to the other members of the coronaviruses family but has major variations in the gene sequence in comparison to earlier sequenced CoVs owing to its incorporation of a polybasic cleavage site that result in augmented pathogenicity and transmissibility (He et al. 2020; Coutard et al. 2020).
The SARS-CoV-2 has an RNA sequence of around 30,000 nucleotides responsible for encoding the whole viral proteome. Entire coding SARS-CoV-2 RNA can be divided into three central regions, non-structural protein (nsp) coding region, structural protein-coding region, and accessory protein-coding region (Wang et al. 2020a, b, c; Andersen et al. 2020; Kim et al. 2020). SARS-CoV-2 S-protein (spike protein), a trimeric class I viral fusion glycoprotein, undergoes a structural rearrangement to aid the fusion of the virus with the membrane of the host cell (Li et al. 2016). The angiotensin-converting enzyme 2 (ACE-2) acts as a receptor for SARS-CoV-2. The virus’s S-protein reacts with the pneumocytes’ ACE-2 with a high affinity, thereby acting as a gateway to the cell (Oudit et al. 2009; Wrapp et al. 2020).
Viral proteases, RNA-dependent RNA polymerase, and viral surface spike protein are among those few of the significant proteins that act as potential drug targets for SARS-CoV-2 based on the earlier therapeutics developed against earlier coronaviruses (Medhi et al. 2020; Nadeem et al. 2020). For battling the SARS-nCoV-2, there is a need to create a quick treatment to curb the disease spread, which could only be achieved by drug repurposing (Tu et al. 2020; Cherian et al. 2020). Drug repurposing is achieved by various computational and experimental techniques (Asai et al. 2020; Pawar et al. 2020). As mentioned, three essential drug targets have been studied extensively using drug repurposing (Elfiky et al. 2020; Fan et al. 2020). Owing to its critical role in pathogenesis and high sequence conservation, the main protease (Mpro, 3CLpro) is regarded as an efficient target for drug design and development (Kruse et al. 2020).
Several existing medications are being evaluated for the treatment of COVID‑19, including antiviral drugs, quinine compounds (Zumla et al. 2016; Zhang et al. 2020). Two broad-spectrum IND having antiviral activity remdesivir (GS-5734) and favipiravir (T-705) and five FDA-approved drugs (penciclovir, ribavirin, nafamostat, nitazoxanide, and chloroquine (C.Q.) were tested against the clinical isolate of SARS-CoV-2 (Koch et al. 2020). Extensive clinical trials of the mentioned drugs help to conclude that remdesivir (GS-5734), an experimental drug developed for the treatment of Ebola hemorrhagic fever caused by a filovirus, and C.Q., an anti-malarial drug, is showing substantial efficacy (Wang et al. 2020a, b, c; Zhou et al. 2020a, b).
Researchers from different fields work in various aspects by applying different approaches to battle against this pandemic (Zeng et al. 2021; Singh et al. 2020). Aiming to contribute towards rapid therapeutic development, we opted for Virtual Screening of currently FDA-approved drugs using the COVID-19 main protease as a drug target. Bromocriptine binds to the active site of ZIKV-NS2B-NS3 protease and could thereby inhibit ZIKV replication (Chan et al. 2017). Previous studies also reported that it inhibits the translation and replication of the dengue virus by binding to NS-3 protease (Kato et al. 2016), revealing that it could have activity against SARS-CoV-2. Ergot alkaloids methylergometrine and methysergide used to treat migraine were studied for repurposing since its congener ergotamine showed significant activity against the different targets SARS-CoV-2 (Mevada et al. 2020). Indispensable cellular serine proteases TMPRSS2 are highly expressed at epithelial cells in human lungs and co-expressed with the SARS-CoV-2 receptor ACE-2 on type II pneumocytes (Donaldson et al. 2001; Paoloni-Giacobino et al. 1997). This regulates the S protein priming and efficiently activates SARS-CoV-2 S protein to induce virus-cell membrane fusion at the cell surface. The inhibition of TMPRSS2 could thereby act as an attractive drug target for the treatment of COVID-19.
Materials and methods
Homology modeling of TMPRSS2
Homology modeling was utilized to generate a valid protein structure by using the sequence of amino acids. The 3-D crystal structure of TMPRSS2 is not established, so we used the SWISS-modeler (Waterhouse et al. 2018; Bienert et al. 2016; Guex et al. 2009) to generate the homology model of TMPRSS2. SWISS-MODEL developed the three different models for the TMPRSS2. Out of the three models, we selected the best model for further molecular docking based on the QMEANS value (Fig. 1a). The homology model can be considered reliable when the target sequence alignment is more than 30 %. The sequence alignment between the developed model of TMPRSS2 and human hepsin TMPRSS1 (5ce 1.1 A) is 33.82 % of sequence similarity and has a resolution of 2.4 Å (Fig. 1c). The model showed a total of 116 similar amino acid residues with the hepsin, out of which 92 residues were in catalytic (Singh et al. 2020) or active site of the generated TMPRSS2 homology model, which enhanced the reliability of this model.
This model was built based upon the template-target alignment by using ProMod3. The geometry of the model was regularized by using the force field. The quality of the model was analyzed by the QMEANS and Generalized Quantum Master Equation (GMQE) value of the model, which was found to be − 1.43 and 0.53, respectively (Fig. 1b). The RAMPAGE server further analyzed the quality of the model. The result found that out of 344 amino acids of the homology model, 92.7 % of the total residues are in favored regions, 6.7 % are in allowed regions, and only 0.6 % are in the outer regions.
The PDB ID was downloaded and used for the docking (Fig. 1a).

a Homology model for TMPRSS2, b local quality estimation with a chart for target by SWISS-Modeler, and c predicted sequence alignment of the model target (TMPRSS2) concerning Human Hepsin TMPRSS1
Selection of ligands and targets
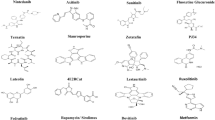
This study selected the FDA-approved drugs, which are semi-synthetic derivatives of a natural ergot alkaloid. These compounds have been studied on the main protease (Mpro), RdRp, and TMPRSS2. The crystal structure of the main protease (6LU7) and RdRp (6M71) were downloaded from the RSCB protein database in PDB format (www.rscb.org). The PDB file of the protein was cleaned with the help of the BIOVIA discovery studio.
Virtual screening and molecular docking
The molecular docking was performed by PyRx version 0.8 Autodock vina (https://pyrx.sourceforge.io/). The protein molecules TMPRSS2, RdRp, and main protease Mpro were loaded into software individually put the macromolecules as fixed. The ligands have been rotatable torsions. The size of box was kept as center_x = − 26.284, center_y = 12.5976 and center_z = 58.9679 for main protease, center_x = 121.4969, center_y = 123.2721 for RdRp and center_z = 127.0716 and center_x = 1.1075, center_y = − 1.3337 and center_z = 15.7311 for TMPRSS2 for docking towards all the ligands with exhaustiveness parameter of 8. The BIOVIA discovery studio analyzed the ligand-protein interaction.
FEP-ABFE approaches
The accelerated FEP-ABFE technique was relied to on the utilization of the RED function. The RED function was designed to automatically add restraints that helped achieve the single-step perturbation to analyze free binding energy and accelerate the FEP-ABPE analysis (Li et al. 2020b). The FEP-ABPE approaches used the 42 λ value (Aldeghi 2016, 2017), but the RED function-based FEP-ABFE can be analyzed and gives the best results in the 16 λ. After molecular docking, the best compound is subjected to the RED function-based FEP-ABFE method. With this function, the compound was simulated against the selected targets up to the 20 λ. The best binding energy was found at 16 λ.
Target and toxicity prediction of ligands
This analysis was necessary to predict possible targets of the selected drugs. The SWISS target prediction server was used for these studies. The toxicity prediction was needed to analyze the concentration of safe drugs for human use (Daina et al. 2019). The toxicity prediction was performed using the pkCSM online database. The drugs’ experimental toxicity is mentioned with the Drug Bank server’s help (David et al. 2017) (https://go.drugbank.com/). The input files of the molecules were submitted in smiles format. This online database gives the AMES toxicity, maximum tolerated dose, hERGI, hERGII, LD50 with liver, and skin toxicity (Pires et al. 2015).
Molecular dynamic and MM-PBSA analysis
Molecular dynamics were executed with a docked structure having minimum energy using GROMACS software (Version 5.1.2) (Berendsen et al. 1995) with CHARMM36-March 2019 (Lee et al. 2014) force field using TIP3P (Boonstra et al. 2016). For the preparation of ligand files, the CHARMM General Force Field server (http://cgenff.umaryland.edu/) was used. PBC was applied by generating a dodecahedron box. An adequate number of Na + or Cl− ions were added for the neutral system. Energy minimization was followed by system equilibration for 100 ps at 300 K using isochoric-isothermal (NVT) equilibration by keeping time step of 2 fs. With the same time step, the isothermal-isobaric ensemble was performed for 100 ps at 300 K. Electrostatic and van der Waals interactions cut-offs for both NVT and NPT were kept at 1.2 nm. For long-range calculating interactions, smooth particle mesh Ewald (PME) method was used. M.D. Simulation of 20,000 ps was performed using the same cut-off, and 20,000 ps trajectories were submitted to MM-PBSA analysis with 20,000 frames for TMPRSS2, RdRp protein, Main protease (Kumari et al. 2014; Baker et al. 2001).
Results
Molecular docking
We have selected five FDA-approved drugs, which are semi-synthetic derivatives of natural ergot alkaloids. The antiviral properties of these compounds have already been established. The compound named bromocriptine was shown to be a potent serine protease that is already reported as an antiviral agent (Kato et al. 2016). Other compounds from the same class that is isolated from the fungus have similar antiviral properties. This study aims to repurpose these compounds against the SARS-CoV-2 protease (Mpro), RdRp, and TMPRSS2 serine protease. These five compounds’ molecular docking study showed good affinity towards the main protease (Mpro), RdRp, and TMPRSS2 serine protease. We have selected a total of three reference compounds, N3, remdesivir, and camostat mesylate, for this study. N3, remdesivir, and camostat mesylate are already established as main protease inhibitors (Mpro), RdRp, and TMPRSS2, respectively (Fig. 2).

Docking score of the drugs against the targets
The bromocriptine showed the highest affinity towards the selected targets. It showed the binding affinity of − 9.6 kcal/mol for the main protease, − 9.3 kcal/mol for the RdRp protein of SARS-CoV-2, and − 8.8 kcal/mol for TMPRSS2 serine proteases. Whereas the reference compounds, i.e., N3 showed − 7.5 kcal/mol for the main protease, remdesivir showed − 8.4 kcal/mol for the RdRp protein of SARS-CoV-2, and camostat mesylate showed − 7.1 kcal/mol for TMPRSS2 serine proteases that are low from the docking score of bromocriptine for all of the targets (Fig. 2). Bromocriptine showed hydrogen bonding with GLY143A, ARG188A, ASN 142 A, and van der Waals interaction with THR190A, GLN192A, ASP187A, HIS164A, SER144A, GLU166A, CYS145A, LEU27A, THR26A, and THR25A residue of main protease Mpro. The N3 showed hydrogen bonding with THR190A, HIS 41 A, GLU166A, GLN189A, GLY143A, THR26A, and van der Waals interaction with TYR54A, ARG188A, ASP187A, HIS172A, ASN142A, SER144A, MET49A, CYS145A, THR25A, LEU27A, LEU167A, and GLN192A (Fig. 3).

3D structure interaction of ligand-protein at the left side and 2D interaction of at right side, a 3D structure interaction of Bromocriptine-Mpro protease, b 2D interaction of Bromocriptine-Mpro protease, c 3D structure interaction of N3-Mpro protease, and d 2D interaction of N3-Mpro protease
Further, bromocriptine showed two conventional hydrogen bonding with the LYS780A, one conventional hydrogen bonding with TYR129A, and one carbon-hydrogen bond with GLN773A. The bromocriptine shows the van der Waals interaction with the ASN138 A, ASP140A, LEU142A, THR141A, CYS139A, ASP135A, HIS 133 A, ASN 781 A, SER 709 A, ASP 711 A, THR710A, and SER772A. Remdesivir showed hydrogen bonding with TYR129A, HIS133A, SER709A, ASN780A, GLY774A, LYS47A, GLN773A, and van der Waals interaction with ASN705A, LYS780A, SER784A, ASP135A, PHE134A, ASP140A, THR141A, LEU142A, CYS139A, ASN138A, ALA706A, THR710A, ASP711A (Fig. 4).

3D structure interaction of ligand-protein at the left side and 2D interaction of at right side, a 3D structure interaction of Bromocriptine-RdRp, b 2D interaction of Bromocriptine-RdRp, c 3D structure interaction of Remdesivir-RdRp, and d 2D interaction of Remdesivir-RdRp
In TMPRSS2, the bromocriptine showed hydrogen bonding with ARG150A, CYS241A, and the van der Waals interaction with GLN159A, ILE452A, GLY153A, ALA243A, and PHE156A. Camostat mesylate showed hydrogen bonding with GLY464A, CYS 465 A, HIS296A, ASP435A, and van der Waals interaction with SER460A, ALA466A, TRP461A, GLY462A, GLY472A, SER436A, VAL473A, THR459A, THR393A, HIS279A, VAL278A, ALA386A, VAL280, GLY439A, SER441A (Fig. 5). The binding energy of bromocriptine was found to be -8.8 kcal/mol, was already established as the serine protease inhibitor (Chan et al. 2017) (Fig. 5).

3D structure interaction of ligand-protein at the left side and 2D interaction of at right side, a 3D structure interaction of Bromocriptine-TMPRSS2, b 2D interaction of Bromocriptine-TMPRSS2, c 3D structure interaction of Camostat mesylate-TMPRSS2, and d 2D interaction of Camostat mesylate-TMPRSS2

Interactive residues of SARS-CoV-2 targets with Bromocriptine and reference compounds
The predicted conventional hydrogen binding residues of bromocriptine and reference compounds with selected COVID-19 protein targets and their respective bond lengths (Å) are mentioned in Fig. 6. The results found that all the bond length was found to be less than 4 Å which concludes that the compounds having good binding with the targets.
Target prediction and toxicity prediction
The pkCSM online database identified the prediction of the toxicity of the drugs. The bromocriptine showed no AMES toxicity, hepatotoxicity, and skin sensitivity (Table 1). None of the compounds interfered with the hERGI inhibitors. The LD50, LOAEL doses are mentioned in Table 1. But the ergot alkaloid, methysergide, and pergolide showed AMES toxicity. The existed experimental toxicity of the drugs is mentioned by using the Drug bank server. The drugs showed nausea, vomiting, diarrhoea, and severe hypotension as common side effects. Bromocriptine is currently used to manage the neurological disease and metabolic disorders such as Parkinson’s, acromegaly, and type-2 diabetes mellitus via (Ozery et al. 2020). Further, the SWISS target prediction server analyzed the target prediction of bromocriptine. The bromocriptine contributes 46 % family A G-protein-coupled receptor, 20 % proteases, 12 % kinases, 8 % cytochrome P450, 6 % enzymes. The SWISS target prediction showed there are chances of protease enzyme inhibition by bromocriptine (Fig. 7).

Target prediction of Bromocriptine
Molecular dynamic and MM-PBSA analysis
A molecular dynamics simulation study of the docked structure was done to analyze the drug’s effect on the Mpro, RdRp, and TMPRSS2. The molecular dynamics were performed up to 20 ns, and results indicate that bromocriptine has an enormous impact on the molecular structure of the Mpro, RdRp, and TMPRSS2 protein. Figure 8 shows the proteins’ RMSD plots (Mpro, RdRp, and TMPRSS2 proteins) and bromocriptine-protein complex. This indicated that bromocriptine stabilizes the protein structures. The RMSD value of bromocriptine-Mpro was less than 0.25 Å, similar to the N3 (Fig. 8a). In the bromocriptine-TMPRSS2, good stabilization started from 0.1 Å throughout the simulation process compared to the camostat mesylate (Fig. 8b). The RMSD plot value of bromocriptine-TMPRSS2 was found to be in the range of 0.2–0.3 Å without fluctuations.
Similarly, the bromocriptine-RdRp complex showed good stabilization throughout the simulation, and the RMSD value was found near 0.2 Å without much fluctuation. The result obtained by bromocriptine showed a similar pattern as that of remdesivir-RdRp complex (Fig. 8c). The RMSD values of bromocriptine towards three different targets indicate that bromocriptine was tightly bound to its active site.

RMSD plot of bromocriptine with a Main protease (Mpro), b TMPRSS2 and c RdRp
These complexes were further analyzed for their interaction with particular residues by molecular dynamic studies. Bromocriptine-Mpro complex had single conventional hydrogen bonding with ARG188A and two conventional carbon-hydrogen bonding with GLY143A before starting the M.D. study. At a 10 ns frame of MDS, the bromocriptine stayed at the same protein pocket with conventional hydrogen bond with the identical residues of the protein, i.e., ARG188A and GLY143A. At 20 ns frame of MDS, the bromocriptine lost its bonding with GLY143A conventional hydrogen bond and formed an additional conventional hydrogen bond with VAL186A with a distance of 1.98 Å in complex stabilization (Fig. 9).

Interaction analysis of bromocriptine bounds to Mpro binding domain during simulation process. a Binding of bromocriptine with Mpro before molecular simulation study, b Binding of bromocriptine with Mpro with molecular simulation study at 10 ns, c binding of bromocriptine with Mpro with molecular simulation study at 20 ns, d 2D interaction of the bromocriptine with Mpro before molecular simulation study, e 2D interaction of the bromocriptine with Mpro with molecular simulation study at 10 ns, f 2D interaction of the bromocriptine with Mpro with molecular simulation study at 20 ns
To shed light on the bromocriptine-RdRp complex, this complex has three conventional hydrogen bonding, two bonding with LYS780A (2.68 and 2.84 Å), and one conventional bonding with TYR129A (2.48 Å) before M.D. study. After the M.D. study at 10 ns, all three conventional hydrogen bonds were lost and formed additional hydrogen bonds with ASP135A and LYS714A. At 20 ns, bromocriptine formed two new hydrogen bonds with CYS 799 A with a distance of 2.72 Å and 2.77 Å. This bonding increased the stability of the complex during MDS (Fig. 10).

Interaction analysis of bromocriptine bounds to RdRp binding domain during simulation process. a Binding of bromocriptine with RdRp before molecular simulation study, b binding of bromocriptine with RdRp with molecular simulation study at 10 ns, c binding of bromocriptine with RdRp with molecular simulation study at 20 ns, d 2D interaction of the bromocriptine with RdRp before molecular simulation study, e 2D interaction of the bromocriptine with RdRp with molecular simulation study at 10 ns, f 2D interaction of the bromocriptine with RdRp with molecular simulation study at 20 ns
The binding mode of bromocriptine with the RBD domain of the TMPRSS2 is elaborated in Fig. 11. It was observed that the TMPRSS2 and bromocriptine-TMPRSS2 protein complex RMSD is very stable, it was found that bromocriptine stayed in the same protein pocket throughout the MDS process. At 10 ns, an additional conventional hydrogen bond was formed with CYS244A with a distance of 2.33 Å, providing stability to the complex. At 20 ns frame, another hydrogen bond formed with THR190A with a bond length of 2.58 Å, and the bond distance of CYS244A become shorter from 2.33 to 2.19 Å. The MM-PBSA residue-based decomposition analysis showed that the THR 190, ALA 243, CYS 244, CYS 241 contributes − 2.30, − 1.814, − 0.619, − 0.854 kcal/mol of binding energy.

Interaction analysis of bromocriptine bounds to TMPRSS2 binding domain during simulation process. a Binding of bromocriptine with TMPRSS2 before molecular simulation study, b binding of bromocriptine with TMPRSS2 with molecular simulation study at 10 ns, c binding of bromocriptine with TMPRSS2 with molecular simulation study at 20 ns, d 2D interaction of the bromocriptine with TMPRSS2 before molecular simulation study, e 2D interaction of the bromocriptine with TMPRSS2 with molecular simulation study at 10 ns, f 2D interaction of the bromocriptine with TMPRSS2 with molecular simulation study at 20 ns
To further analyze the interaction between targets’ active sites and bromocriptine, the root means square fluctuations (RMSF) studies were performed. RMSF study of the bromocriptine with Mpro shows slight variation in comparison with Mpro protein. The residues of Mpro active site that binds to the bromocriptine showed significantly less average RMSF value, i.e., less than 0.25 nm. We have found maximum fluctuation in THR 169 and ASN 238 residues with RMSF values of 0.206 and 0.288 nm (Fig. 12a).
Similarly, in the bromocriptine-TMPRSS2 complex, the fluctuation was also observed in amino acid residue at the protein’s bromocriptine binding site, and the RMSF value was found to be around 0.45 Å. The residues involved in the fluctuation are ASP 175, ASN 218, LYS 340, GLY 370, and PRO 422, with average values of 0.2147, 0.4497, 0.408, 0.2919, and 0.1999 nm, respectively. It is assumed that very low b-factor in the region owing to the structure confirmation (Fig. 12b).
In the case of RdRp, the RMSF study showed fluctuation in PRO112, LYS 160, LEU 261, ASN 911 amino acid residues with average RMSF values of 0.308, 0.2423, 0.4974, and 0.4162 nm, respectively (Fig. 12c).

RMSF plot of bromocriptine with a main protease (Mpro), b TMPRSS2 and c RdRp protein
Furthermore, we examined the solvent-accessible surface area (SASA) to inspect the hydrophilic and hydrophobic residues of the control targets and bromocriptine docked target complex. M.D. simulation-based decrease in the average percentile value in SASA for the active pocket of proteins indicates that ligand is reliable to penetrate the core of protein (Morris et al. 2019). In this study, the SASA plot of bromocriptine-Mpro has slight fluctuation throughout the M.D. process, the average value of this complex and Mpro apo-protein was found to be 168.25 and 169.02 nm2 (Fig. 13a). The bromocriptine-TMPRSS2 and TMPRSS2 showed the plateau after 5 ns and stayed the same up to 20 ns of M.D. simulation with the SASA value of 188.27 and 186.65 nm 2 respectively (Fig. 13b). The third complex, bromocriptine-RdRp, showed stability up to 10 ns of the M.D. process. After that, the complex had some fluctuation but regained stability after 15 ns of M.D. process. The bromocriptine-RdRp complex and RdRp value’s average SASA value was 469.48 and 469.28 nm2 (Fig. 13c).

SASA plot of bromocriptine with a main protease (Mpro), b TMPRSS2 and c RdRp protein
The Radius of gyration (Rg) indicates the compactness, shape, and folding of the protein and ligand-protein complex. The system with a higher number of Rg shows higher structure compactness. Figure 14 represents the Rg plots of bromocriptine with the Mpro, RdRp, and TMPRSS2. Plots revealed that bromocriptine-protein complexes have more compactness as compared to the protein control. The bromocriptine-Mpro showed the plateau from the beginning of molecular dynamics upto 10,000 ps. The Mpro protein and bromocriptine-Mpro complex were stabilized between 2.20 and 2.5 nm, respectively (Fig. 14a). The bromocriptine-TMPRSS2 and TMPRSS2 protein started the plateau from 10 to 15 ns. The Rg value of the bromocriptine-TMPRSS2 and TMPRSS2 was found to be 2.17 ± 0.3 (Fig. 14b). In the case of bromocriptine-RdRp shows the plateau up to 8 ns, the RdRp protein and bromocriptine-RdRp complex having the Rg value between 3.0 and 3.05 nm (Fig. 14c).

Rg plot of bromocriptine with a Mpro, b TMPRSS2, c RdRp protein
MM-PBSA method was performed on the entire three ligand-protein complexes for screening the binding free energy of the bromocriptine towards the Mpro, RdRp, and TMPRSS2. The binding free energy calculation was performed up to 20,000 ps on the M.D. trajectories. This method’s analysis of the free binding energy is more favorable than the ligand-protein complex’s docking score. Bromocriptine-TMPRSS2 showed the highest binding energy of − 18.77 kcal/mol, followed by the bromocriptine-Mpro with − 17.85 kcal/mol, and bromocriptine-RdRp has the least binding energy of − 6.30 kcal/mol (Fig. 15).

MM-PBSA based binding energy calculation
FEP-ABFE approaches
RED function-based FEP-ABPE results calculated for the bromocriptine against the Mpro, RdRp, and TMPRSS2 targets. This method helps encounter systematic errors, and the results showed that the best binding energy was found to be at 16 λ. We use BAR analysis, which provides a simple path to perform all calculations and produces various error analysis procedures. The free energy of solvation of the bromocriptine-Mpro, bromocriptine-RdRp bromocriptine- TMPRSS2 was found to be − 5.114 ± 0.038, − 4.343 ± 0.123, and − 5.195 ± 0.036 kcal/mol. The calculation’s visual comparison segments are mentioned in the supplementary figures (Figures S1, S2, and S3).
Conclusion
In the present study, we analyzed the structures of SARS-CoV-2 Mpro, RdRp and TMPRSS2 co-crystallized with five different inhibitors possessing antiviral activity. We explored their binding affinity, which revealed few potential drugs that can be considered for repurposing. Our analysis also predicted toxicity profiles of all the ligands (Table 1) considering AMES toxicity, hepatotoxicity, skin sensitivity, and hERG inhibition for assessing potential cardiotoxicity. Bromocriptine, methysergide, methylergometrine, and cabergoline showed good docking scores when docked with the mentioned SARS-CoV-2 proteins. Among these compounds, bromocriptine is a promising candidate to inhibit SARS-CoV-2 replication. It is bound with a high affinity to all the receptors with the binding affinity of − 9.6 kcal/mol towards the Mpro, − 9.3 kcal/mol for the RdRp protein, − 8.8 kcal/mol against the TMPRSS2, and showed no toxicity in all domains of assessment.
In conclusion, the accelerated FEP-ABFE show 5 different method TI, TI-CUBIC, DEXP, IEXP, BAR results for energy calculation methods show a bit different result at state 12–13, 13–14, 14–15, at rest they line up good. We used the BAR for reporting results as it was better as compared to other methods. The free energy of solvation of the bromocriptine-Mpro, bromocriptine-RdRp bromocriptine- TMPRSS2 was found to be − 5.114 ± 0.038, − 4.343 ± 0.123, and − 5.195 ± 0.036 kcal/mol. These predictions have been performed, resulting in the bromocriptine’s incredible accurate binding energy with selected targets. The molecular dynamic studies and MM-PBSA study also suggested that bromocriptine can be the best candidate for the Main protease (Mpro), TMPRSS2, and RdRp. Furthermore, previous studies showed that bromocriptine inhibits protease activity of Zika virus (Chan et al. 2017) and the translation or replication steps in the dengue virus life cycle (Kato et al. 2016), confirming our approach for utilization of bromocriptine against SARS-CoV-2 infection. Since these FDA-approved compounds’ safety pharmacology is already established, it would be easy to repurpose them to meet the grave need for treatment of COVID-19.
References
Al-Tawfiq J (2020) Asymptomatic coronavirus infection: MERS-CoV and SARS-CoV-2 (COVID-19). Travel Med Infect Dis 35:101608
Aldeghi M, Heifetz A, Bodkin MJ, Knapp S, Biggin PC (2016) Accurate calculation of the absolute free energy of binding for drug molecules. Chem Sci (Camb) 7:207–218
Aldeghi M, Heifetz A, Bodkin MJ, Knapp S, Biggin PC (2017) Predictions of ligand selectivity from absolute binding free energy calculations. J Am Chem Soc 139:946–957
Andersen K, Rambaut A, Lipkin W, Holmes E, Garry R (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452
Asai A, Konno M, Ozaki M et al (2020) COVID-19 drug discovery using intensive approaches. Int J Mol Sci 21:2839
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041
Berendsen HJ, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56
Bienert S, Waterhouse A, de Beer T et al (2016) The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res 45:D313–D319
Boonstra S, Onck PR, van der Giessen E (2016) CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J Phys Chem B 120:3692–3698
Chan JF-W, Chik KK-H, Yuan S et al (2017) Novel antiviral activity and mechanism of bromocriptine as a Zika virus NS2B-NS3 protease inhibitor. Antiviral Res 141:29–37
Chan J, Lau S, Woo P (2013) The emerging novel Middle East respiratory syndrome coronavirus: The “knowns” and “unknowns”. J Formos Med Assoc 112:372–381
Chen N, Zhou M, Dong X et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395:507–513
Cherian S, Agrawa M, Basu A et al (2020) Perspectives for repurposing drugs for the coronavirus disease 2019. Indian J Med Res 151:160–171
Coutard B, Valle C, de Lamballerie X et al (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176:104742
Daina A, Michielin O, Zoete V (2019) Swiss Target Prediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 47:357–364
David S, Wishart YD, Feunang AC, Guo EJ, Lo A, Marcu, Jason R, Grant T, Sajed D, Johnson C, Li Z, Sayeeda N, Assempour I, Iynkkaran Y, Liu A, Maciejewski N, Gale A, Wilson L, Chin R, Cummings D, Le (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46(D1):D1074–D1108
Donaldson S, Hirsh A, Li D et al (2001) Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem 277:8338–8345
Elfiky A (2020) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci 253:117592
Fan H, Wang L, Liu W et al (2020) Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin Med J 133:1051–1056
Guex N, Peitsch M, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30:S162–S173
He F, Deng Y, Li W (2020) Coronavirus disease 2019: What we know? J Med Virol 92:719–725
Kato F, Ishida Y, Oishi S et al (2016) Novel antiviral activity of bromocriptine against dengue virus replication. Antiviral Res 131:141–147
Khan S, Zia K, Ashraf S, Uddin R, Ul-Haq Z (2020) Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn 1–10
Kim D, Lee J, Yang J et al (2020) The architecture of SARS-CoV-2 transcriptome. Cell 181:914-921.e10
Koch S, Pong W (2020) First up for COVID-19: nearly 30 clinical readouts before end of April. BioCentury Inc
Kruse R (2020) Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Res 9, 72
Kumari R, Kumar R (2014) Open Source Drug Discovery Consortium, Lynn A. g_mmpbsaî¸ A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962
Lee S, Tran A, Allsopp M et al (2014) CHARMM36 united atom chain model for lipids and surfactants. J Phys Chem B 118:547–556
Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3:237–261
Li Q, Guan X, Wu P et al (2020) Early transmission dynamics in Wuhan, China, of Novel Coronavirus–infected pneumonia. N Engl J Med 382:1199–1207
Li Z et al (2020b) Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. PNAS. 117:27381–27387
Lupia T, Scabini S, Mornese Pinna S et al (2020) 2019 novel coronavirus (2019-nCoV) outbreak: a new challenge. J Glob Antimicrob Resist 21:22–27
Medhi B, Prajapat M, Sarma P et al (2020) Drug for corona virus: A systematic review. Indian J Pharmacol 52:56
Mevada V, Dudhagara P, Gandhi H et al (2020) Drug Repurposing of Approved Drugs Elbasvir, Ledipasvir, Paritaprevir, Velpatasvir, Antrafenine and Ergotamine for Combating COVID19. ChemRxiv. Preprint
Morris KF, Billiot EJ, Billiot FH, Ingle JA, Krause KB, Lewis CR, … Fang Y (2019) Using molecular dynamics simulations to identify the key factors responsible for chiral recognition by an amino acid-based molecular micelle. J Dispersion Sci Technol 40:716–727
Nadeem M, Zamzami M, Choudhry H et al (2020) Origin, potential therapeutic targets and treatment for coronavirus disease (COVID-19). Pathogens 9:307
Oudit G, Kassiri Z, Jiang C et al (2009) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest 39:618–625
Ozery M, Wadhwa R (2020) Bromocriptine. StatPearls [Internet]
Paoloni-Giacobino A, Chen H, Peitsch M, Rossier C, Antonarakis S (1997) Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 44:309–320
Pawar A (2020) Combating devastating COVID-19 by drug repurposing. Int. J. Antimicrob. Agents, 105984
Pires D, Blundell T, Ascher D (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072
Singh R, Gautam A, Chandel S et al (2020) Protease inhibitory effect of natural polyphenolic compounds on SARS-CoV-2: an in silico study. Molecules 25:4604
Smith M, Smith JC (2020) Repurposing Therapeutics for COVID-19: supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. ChemRxiv.; Preprint
Tu Y, Chien C, Yarmishyn A et al (2020) A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci 21:2657
Wang M, Cao R, Zhang L et al (2020c) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271
Wang H, Li X, Li T et al (2020a) The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur J Clin Microbiol Infect Dis, 1–7
Wang K, Chen W, Zhou YS et al (2020b) SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv (preprint)
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Wrapp D, Wang N, Corbett K et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–1263
Wu Z, McGoogan J (2020) Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China. JAMA 323:1239
Zeng W, Gautam A, Huson DH (2021) On the Application of Advanced Machine Learning Methods to Analyze Enhanced, Multimodal Data from Persons Infected with COVID-19. Computation 9:4
Zhang L, Lin D, Sun X et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, eabb3405
Zhou D, Dai S, Tong Q (2020a) COVID-19: a recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J Antimicrob Chemother 75:1667–1670
Zhou F, Yu T, Du R et al (2020b) Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062
Zumla A, Chan J, Azhar E, Hui D, Yuen K (2016) Coronaviruses—drug discovery and therapeutic options. Nat Rev Drug Discov 15:327–347
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, R., Gautam, A., Chandel, S. et al. Computational screening of FDA approved drugs of fungal origin that may interfere with SARS-CoV-2 spike protein activation, viral RNA replication, and post‐translational modification: a multiple target approach. In Silico Pharmacol. 9, 27 (2021). https://doi.org/10.1007/s40203-021-00089-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-021-00089-8