
Abstract
Purpose
Vortioxetine an anti-depressant FDA-drug recently reported showing better in vitro efficacy against SARS-CoV-2.
Methods
In this study, we have synthesized ten new derivatives having alkenes, alkynes, benzyl, aryl, and mixed carbamate at the N-terminal of vortioxetine. Then the binding energy and interactions with the crucial amino acid residues in the binding pocket of main protease (Mpro) of SARS-CoV-2, of reported and ten newly synthesized vortioxetine derivatives (total thirty-one) in comparison with remdesivir are analyzed and presented in this paper.
Results
Based on the docking scores predicted by ADV and AD, most vortioxetine derivatives showed better binding efficiency towards Mpro of SARS-CoV-2 in comparison with remdesivir (an EUA approved drug against SARS-CoV-2 Mpro) and vortioxetine.
Conclusion
This study shows that some vortioxetine derivatives can be developed into promising drugs for COVID-19 treatment.
Similar content being viewed by others

Introduction
The world is now facing a serious health crisis condition due to coronavirus disease (COVID-19) since December 2019 [1]. The causative agent for COVID-19 was severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). SARS-Cov-2 belongs to genus β-coronavirus (order Nidovirales; family Coronaviradae) and is a non-segmented, ( +)-sense, enveloped single-stranded RNA virus [2]. It consists of genome length ranging from 26 to 32 kb in length [3]. Two proteases that facilitate the processing of functional proteins of SARS-CoV-2 are the 3C-like protease (3CLpro) and the papain-like protease (PLpro) [4]. 3CLpro executes proteolytic cleavages at the maximum number of sites (11 sites) within the polyprotein hence it is also termed as the main protease (Mpro) [5]. Mpro has a molecular weight of 33.8 kDa and is reported to be a cysteine protease. Within SARS-CoV-2, each protomer of Mpro protein are homodimer consisting of three domains – domain I, domain II and domain III [6]. The catalytic site/active site/substrate-binding site of SARS-CoV-2 Mpro are located at the cleft of domains I and II, comprised of His-Cys catalytic dyad (cysteine-145 and histidine-41 moieties) [7]. Here cysteine-145 acts as a common nucleophile and plays a major role in the proteolytic functioning of Mpro. Hence Mpro emerged as an important drug target against SARS-CoV-2, since it plays a vital role in polyprotein processing, virus maturation and the absence of similar protease in humans also makes it a perfect choice. Recently X-ray crystallography structure of Mpro, co-crystallized with an inhibitor N3 (PDB ID: 6LU7) had been reported by Jin et al. [6]. In last six months few vaccines had also been developed against SARS-CoV-2 which reduces the rate of infection and thus mortality [8]. Researchers are in continuous search for suitable alternatives like nanoparticles, small molecule drugs [9], and antibodies for the treatment of COVID-19 [10,11,12,13]. In present emergent situation small molecules drugs like remdesivir and favipiravir are now used for the treatment of COVID-19 [14]. Remdesivir is a known antiviral drug, now using in the treatment of COVID-19 which acts by inhibiting SARS-CoV-2 Mpro responsible for polyprotein processing and virus maturation [15].
Many research laboratories around the world are now in continuous search for specific antiviral drugs which can be repurposed successfully for the therapeutic cause without much side-effects [16]. In addition, several other antibiotics, antibodies, NSAIDs and steroids are now been tried for the treatment of COVID-19 and are currently in clinical trials [17]. Fluoxetine, an anti-depressant FDA-drug recently reported as a possible therapeutic agent against COVID-19 (Fig. 1) [18, 19]. Initial data of studies are also interesting as it shows mixed effects against SARS-CoV-2. Another FDA-approved antidepressant drug vortioxetine also showed inhibition of SARS-CoV-2 spike protein mediated cell fusion [20]. It was also reported that vortioxetine in combination therapy with other drugs like clomifene, asenapine and chloroquine showed better efficiency in VSV-SARS-CoV-2-Sdel18 pseudovirus model as well as in authentic SARS-CoV-2 assay [21]. Initially the main option to the researchers to combat the COVID-19 was repurposing of existing drugs but now it is high time to search for new selective molecules to treat the disease. Hence, we envisage the idea of developing vortioxetine derivatives for the therapeutic treatment against this disease. Vortioxetine and its different derivatives are known to possess different activities like immunomodulatory [22], antibacterial [23], antifungal, antioxidant, and anti-inflammatory agents [24,25,26]. Initially, a docking study between Mpro of SARS-CoV-2 and remdesivir was performed. Based on the binding pocket understanding and key features from remdesivir we have used vortioxetine and its reported derivatives as a tool compound to explore their binding interactions with Mpro.

Chemical structure of Remdesivir, Fluoxetine and Vortioxetine (1)
Molecular understanding of vortioxetine helps in designing of more analogues which then could interact in a better way within the binding pocket of Mpro. Then based on binding energy and interaction profile, eight vortioxetine derivatives based on various functionality were synthesized. Then the binding energy and interactions within the binding pocket of Mpro of previously reported and newly synthesized vortioxetine derivatives were analyzed and presented in this paper. The docking score of compounds 15, 17, 19, 21, 26, 28, 29, and 32 predicted by Autodock Vina (ADV) and Autodock 4.2 (AD) against SARS-CoV-2 Mpro were superior to the known inhibitors Remdesivir and Vortioxetine. This study could give a guideline for designing future vortioxetine based drug molecules for inhibiting polyprotein replication and virus maturation by inhibiting SARS-CoV-2 Mpro as a potential target for the treatment of COVID-19.
Result and Discussion
Design and synthesis of vortioxetine derivatives
Initially, remdesivir and vortioxetine (1) were docked to Mpro of SARS-CoV-2, and later additional known derivatives with diverse functionality were studied depending on the initial results (Fig. 2). For more variations, derivatives with different functional groups were designed followed by synthesis and then characterization for structural confirmation. Furthermore, molecular docking studies were carried out for these newly synthesized molecules.

Structures of known derivatives of vortioxetine
Starting material vortioxetine (1) was synthesized by following the previously reported method [23]. The derivatives of vortioxetine were synthesized by base mediated substitution reaction. For this project variations in the N–H terminal were achieved by alkylation, Buchwald coupling, and mixed carbamate formation shown in Fig. 3.

Scheme for synthesis of new vortioxetine derivatives with reaction conditions and isolated yields
To identify the binding interaction inside the binding pocket of Mpro, compound 2–22 with five different types of substitutions at the N-terminal of vortioxetine (acyl sulphonyl, alkyl, benzyl, and / functionalized alkyl group) was initially investigated (Fig. 2). By employing a strong base at ambient condition, the newly designed compounds 23–25 were synthesized from their corresponding halides (Fig. 3). To introduce heteroaromatic groups, the Buchwald reaction was used in conjunction with the Pd2(dba)3 reagent to create compound 26–27. It was well documented that carbamide functionalities sometimes enhance the efficacy of the compound [27]. therefore, from commercially available aryl isocyanate compound 28 and compound 29 were synthesized with moderate yields. Similarly, reaction with corresponding alkyloxy carbonyl chloride at ambient condition gave carbamate functionalized derivative 30. The benzyl derivatives 31 and 32 were synthesized from their corresponding bromides following previously mentioned conditions in moderate yields [24]. All the products were synthesized in low to moderate yields, purified using column/combiflash chromatography, and characterised using 1H-NMR, 13C-NMR, and high-resolution mass spectroscopy. The docking investigation was later conducted using the structures of newly synthesized scaffolds.
Binding mode for known drug Remdesivir and vortioxetine
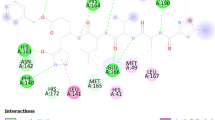
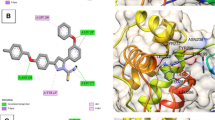
In the beginning, we had carried out molecular docking studies of remdesivir and vortioxetine (1) with the crystal structure of SARS-CoV-2 Mpro (PDB ID: 6LU7). The 2D-interaction and docking poses of both compounds are shown in Fig. 4. Our docking studies revealed that remdesivir forms hydrogen bond interactions with HIP-41, and PHE-140. Additionally, π-anion interaction with HIP-41, MET-49, GLU-166, π-alkyl interaction with MET-49 and carbon-hydrogen interactions HIP-41 and LEU-141 (Fig. 4A). On the other hand, vortioxetine showed π-sulphur interaction with MET-49, π-alkyl interaction with CYS-145, MET-165, and van der Waals interaction with GLN-189 (Fig. 4B). The binding affinity of Remdesivir predicted by Autodock Vina (ADV) and Autodock 4.2 (AD) were ‒7.0 and ‒7.14 kcal/mol, respectively. Similarly, the binding affinity of Vortioxetine predicted by ADV and AD were ‒6.3 and ‒7.41 kcal/mole, respectively. Visual inspection of binding mode of vortioxetine within the binding pocket of Mpro revealed that there is still room for designing stronger Mpro binders by adding more functionality to the piperazine ring (Fig. 4E and F).

The 2D ligand interaction diagram of (A) Remdesivir, (B) Vortioxetine; binding pose of (C, D) Remdesivir (yellow) and (E, F) Vortioxetine (yellow) in the active site of SARS-CoV-2 Mpro
Designing of compounds based on understanding from Remdesivir and vortioxetine binding
Furthermore, molecular docking studies of synthesized derivatives of vortioxetine (compound 2–32) were also performed to understand their binding affinities towards SARS-CoV-2 Mpro. From reported works of literature, a library of compounds 2–22 were selected based on different functionality along with a set of newly synthesized compounds 23–32. All compounds along with their docking score predicted by ADV and AD are listed in Table 1.
Molecular docking studies revealed that most of the designed analogues showed better binding as compared to Remdesivir (-7.0 kcal/mol) and the co-ligand N3 (-7.4 kcal/mol) predicted by ADV [28]. The binding affinities of hits predicted by ADV ≤ ‒7.0 are considered as better inhibitors. ADV predicted binding affinities of hits 15, 17, 19, 26, 28, and 32 would have higher than co-ligand. The binding affinity predicted by ADV and binding energy predict by AD of the designed analogues 15, 17, 19, 21, 26, 28, 29 and 32 were better than the standard Mpro inhibitor Remdesivir. The 2D interactions of the eight best hits depicted in Fig. 5 and their docking score predicted by both the ADV and AD are found in Table 1. The docked binding poses in the 3D surface topology of 15, 17, 19, 21, 26, 28, 29, 32 (green color) superposed on docked pose of known inhibitor remdesivir (yellow color) in the active site of SARS-CoV-2 Mpro are shown in Fig. 6. The binding pose of best hits 15, 17, 19, 21, 26, 28, 29 and 32 was similar to the Remdesivir and adopts a similar binding orientation in the active site. In the active site of SARS-CoV-2 Mpro the residue CYS-145 and HIS-41 acts as catalytic dyad [29]. The selected best hits 15, 17, 21, 26, 28, 29 and 32 exhibited π-anion interactions with CYS-145 and 19 showed hydrogen bonding interaction with CYS-145. Molecular dynamics (MD) simulations of peptide-like drug candidates with SARS-CoV-2 Mpro showed that the crucial amino acid residues for inhibition of Mpro were HIS-41, GLY-143, and GLU-166 [30]. The hit 17 showed hydrogen bonding interactions with HIS-41 and π-anion interaction with GLU-166. The other hit 19 exhibited hydrogen bonding interactions with GLY-143 and π-π T-shaped interactions with GLU-166. The other two hits 28 and 29 interact with either GLY-143 or GLU-166. Therefore, hits 17, 19, 28 and 29 are the best inhibitors concerning inhibition of crucial interacting amino acid residues.

2D ligand interaction diagram best hits (15, 17, 19, 21, 26, 28, 29, and 32 in the active site of SARS-CoV-2 Mpro


Binding poses in the 3D surface topology of selected hits 15, 17, 19, 21, 26, 28, 29, 32 (green color) and remdesivir (yellow color) in the active site of SARS-CoV-2 Mpro (A: Mpro with the active site, B: enlarged binding site)
The molecular weight of all the hits was ≤ 500, the number of hydrogen bond donors ≤ 5, the number of H-bond acceptors ≤ 10. The value of MLOGP ≤ 4.5 [31] for drug-likeness. The MLOGP value for hits 15, 19, 21 and 29 lie in the acceptable range. The topological polar surface area (TPSA) of all the hits were less than 130 and the acceptable range is 20–130 Å2 [32]. The range of the number of rotatable bonds for drug-like molecules should be 0‒9. The range of rotatable bonds of all the selected hits were ≤ 9.0. The range of iLOGP value for the drug-like molecule is -2–10 [33] here for all the hits this value lies in the acceptable range. Gastrointestinal absorption (GI) of all the selected hits were high except 17. The predicted ADME results showed that most of the hits are drug-like. The ADME parameters are shown in Table 2. The synthetic accessibility of all the hits are also good.
Conclusions
In conclusion, most vortioxetine derivatives showed better binding efficiency towards Mpro of SARS-CoV-2 in comparison with remdesivir (an EUA approved drug against SARS-CoV-2 Mpro) and vortioxetine (1). Based on the docking scores predicted by ADV and AD, compound 15, 17, 19, 21, 26, 28, 29, and 32 showed more binding affinity than remdesivir. The hits 17, 19, 28 and 29 are the best inhibitors concerning inhibition of crucial interacting amino acid residues. Among different functionalities, the most efficient is with benzyl derivatives (15 and 17). The predicted ADME results revealed that most of the compounds are drug-like. These four compounds (17, 19, 28 and 29) along with some newly designed derivatives can be screened in future for the model in vitro and in vivo studies. Based on those data prospective vortioxetine derived drugs against SARS-CoV-2 can be developed.
Materials and methodology
Designing of different analogues and synthesis for exploring structure-activity relationship (SAR)
Commercially available analytical grade solvents and reagents were purchased from commercial suppliers and were used without any further purification unless otherwise mentioned. Thin-layer chromatography (TLC) was performed for monitoring progress of reaction using commercially available Merck 60 F254 silica gel plate and visualized under UV light, and/or by spraying with freshly prepared phosphomolybdic acid (PMA) in methanol, followed by charring at high temperature. For purification of the crude compounds, column chromatography was performed on silica gel (100–200 mesh) or by using combiflash. All the 1H NMR and 13C NMR spectra were recorded at 25 °C using chloroform-d(CDCl3) or DMSO-D6 as deuterated solvents with tetramethylsilane (TMS) as an internal standard. The multiplicity of the reported peaks singlet, broad singlet, doublet, triplet, quadruplet and multiplet (or unwell-resolved signals) are denoted by s, br. s, d, t, q, and m respectively. All chemical shifts are reported in ppm (δ) and coupling constants (J) are in hertz (Hz). Mass Spectrometry (MS) data was recorded for unknown compounds 23–32 on Qtof-micro quadruple mass spectrophotometer. Elemental microanalyses were performed on elemental analyzer model flash 2000 thermo fisher for all new compounds.
General procedure for the synthesis of 23–32
New vortioxetine derivatives were synthesized according to following procedures. The synthetic procedure was modified for the synthesis of the vortioxetine derivatives. To a stirred solution of vortioxetine (1) (1 eq) in dry tetrahydrofuran (THF) (10 mL) was added sodium hydride (2 eq) at 0 °C. Contents were stirred at same temperature for 20 min, and then added alkyl/aryl/acyl/sulfonyl halide (1.5 eq) drop wise. The reaction mixture was stirred at room temperature until the reaction completes. The reactions were quenched using cold water and extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with brine, dried over anhydrous sodium sulphate and evaporated under reduced pressure. Crude product was purified by silica gel column chromatography to afford pure compound 23–25, 30–32.
1-(2-((2,4-dimethylphenyl)thio)phenyl)-4-vinylpiperazine, (23)
Yield: 52%; Off white solid, Rf: 0.8, AcOEt: Hexane (1:9), 1H-NMR (CDCl3, 300 MHz) δ 7.29 (d, J = 7.8 Hz, 1H), 7.16–6.73 (m, 5H), 6.39 (d, J = 7.5 Hz, 1H), 5.88–5.79 (m, 1H), 5.17–5.08 (m, 2H), 3.02–3.00 (m, 4H), 2.59–2.56 (m, 4H), 2.26 (s, 3H), 2.22 (s, 3H); 13C-NMR (CDCl3, 75 MHz) δ: 149.15, 142.39, 139.09, 136.19, 134.87, 134.56, 131.58, 127.70, 126.02, 125.36, 124.24, 119.77, 118.13, 61.79, 53.40, 51.49, 21.11, 20.51; MS (m/z): 325.18 [M + H]+. Anal. Calcd for C20H24N2S: C, 74.03; H, 7.46; N, 8.63; Found C, 74.21; H, 7.27; N, 8.48.
1-(2-((2,4-dimethylphenyl)thio)phenyl)-4-(prop-2-yn-1-yl)piperazine, (24)
Yield: 48%; Off White solid, Rf: 0.75, AcOEt: Hexane (1:9), 1H-NMR (CDCl3, 300 MHz) δ 7.29 (d, J = 7.8 Hz, 1H), 7.05–6.74 (m, 5H), 6.41 ( d, J = 7.8 Hz, 1H), 3.28 (s, 2H), 3.06–3.04 (m, 4H), 2.71–2.69 (m, 4H), 2.26 (s, 3H), 2.23 (s, 3H), 2.20 (m, 1H); 13C-NMR (CDCl3, 75 MHz) δ: 149.07, 142.47, 139.19, 136.25, 134.62, 131.69, 128.02, 127.81, 126.19, 125.48, 124.42, 119.88, 78.93, 73.34, 52.44, 51.43, 46.96, 21.22, 20.63; MS (m/z): 337.18 [M + H]+. Anal. Calcd for C21H24N2S: C, 74.96; H, 7.19; N, 8.33; Found C, 74.77; H, 7.25; N, 8.18.
1-(but-2-en-1-yl)-4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazine, (25)
Yield: 29%; Light brown sticky liquid, Rf: 0.7, AcOEt: Hexane (1:9), 1H-NMR (CDCl3, 300 MHz) δ 7.28 (d, J = 7.8 Hz, 1H), 7.05–6.72 (m, 5H), 6.39 (d, J = 7.5 Hz, 1H), 5.60–5.44 (m, 2H), 3.03–2.93 (m, 6H), 2.59–2.56 (m,4H), 2.26 (s, 3H), 2.22 (s, 3H), 1.60 (d, 3H); 13C-NMR (CDCl3, 75 MHz) δ: 149.30, 142.43, 139.13, 136.21, 134.61, 131.64, 129.52, 128.14, 127.76, 126.53, 126.17, 125.46, 124.30, 119.87, 60.90, 53.49, 53.38, 21.16, 20.56, 17.81; MS (m/z): 353.55 [M + H]+; Anal. Calcd. for C22H28N2S: C, 74.95; H, 8.01; N, 7.95; Found: C, 74.77; H, 7.88; N, 7.91.
Benzyl 4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazine-1-carboxylate, (30)
Yield: 69%; White solid, Rf: 0.9, AcOEt: Hexane (1:9), 1H-NMR (CDCl3, 300 MHz) δ 7.30–7.24 (m, 6H), 7.16–6.77 (m, 5H), 6.44 (d, J = 7.8 Hz, 1H), 5.09 (s, 2H), 3.63–3.60 (m, 4H), 2.95–2.93 (m, 4H), 2.27 (s, 3H), 2.23 (s, 3H) 13C-NMR (CDCl3, 75 MHz) δ;155.34, 148.80, 142.23, 139.18, 136.71, 136.01, 134.59, 131.65, 128.46, 127.96, 127.85, 127.75, 126.33, 125.49, 124.65, 119.85, 67.12, 51.48, 44.32, 21.12, 20.52; MS (m/z): 433.19 [M + H]+; Anal. Calcd. for C26H28N2O2S: C, 72.19; H, 6.52; N 6.48; Found: C, 72.01; H, 6.60; N 6.37.
1-(4-bromobenzyl)-4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazine, (31)
Yield: 51%; White solid, Rf: 0.3, AcOEt: Hexane (1:9), 1H-NMR (DMSO-d6, 400 MHz) δ 7.53 (dd, J = 8.4 Hz, 2H), 7.32–7.29 (m, 3H), 7.21 (s, 1H), 7.13–7.06 (m, 3H), 6.90–6.88 (m, 1H), 6.38–6.36 (dd, J = 8.0 Hz, 1H), 3.52 (s, 2H) 2.97 (m, 4H), 2.54–5.52 (m, 4H), 2.32 (s, 3H), 2.22 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ; 149.18, 142.46, 139.20, 137.14, 136.25, 134.60, 131.66, 131.38, 130.99, 127.99, 127.79, 126.11, 125.44, 124.32, 120.96, 119.82, 62.42, 53.51, 51.55, 21.20, 20.60; MS: (m/z) 469.00 [M + H]+.
1-(4-bromo-2-fluorobenzyl)-4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazine, (32)
Yield: 55%; White solid, Rf: 0.4, AcOEt: Hexane (1:9), 1H-NMR (DMSO-d6, 400 MHz) δ 7.52 (dd, J = 8.8 Hz, 1H), 7.51–7.40 (m, 2H), 7.29 (dd, J = 8.0 Hz, 1H), 7.20 (s, 1H), 7.11–7.06 (m, 3H), 6.88 (m, 1H), 6.39–6.37 (dd, J = 8.0 Hz, 1H), 3.58 (s, 2H), 2.97 (m, 4H), 2.54–2.52 (m, 4H), 2.31 (s, 3H), 2.21 (s, 3H) 13C-NMR (CDCl3, 100 MHz) δ; 149.18, 142.39, 139.15, 136.16, 134.58, 132.81, 132.76, 131.65, 128.01, 127.78, 127.28, 127.24, 126.20, 125.46, 124.33, 124.03, 123.88, 121.12, 121.03, 119.85, 119.10, 118.84, 54.89, 53.24, 51.52, 21.19, 20.59; MS (m/z): 484.95 [M + H]+, Anal. Calcd for C25H26BrFN2S: C, 61.85; H, 5.40; N, 5.77; Found: C, 61.85; H, 5.40; N, 5.77.
Cesium carbonate (3 eq), and xantphos (0.05 eq) were added to a solution of vortioxetine (1) (1 eq), aryl bromide (1.5 eq) in 1,4 dioxane (8 mL) solvent. For 20 min, inert argon gas was purged through the reaction mixture, followed by catalytic amounts of Pd2(dba)3(10 mol%) being added. Then the reaction mixture was heated at 110 °C for 12 h. The crude reaction mixture was passed through celite bed and washed with ethyl acetate (50 mL). The organic layer was diluted with water (20 mL), washed with brine, separated, dried over anhydrous Na2SO4 and concentrated to get crude sticky liquid material. Column chromatography (30% ethyl acetate in hexanes) of the crude extract yielded the product as a white solid.
4-(4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazin-1-yl)quinoline (26)
Yield: 44%; Off-White Solid. Rf: 0.4 (AcOEt: Hexane (3:7), 1H-NMR (DMSO-d6, 400 MHz) δ 8.72 (d, J = 4.8 Hz, 1H); 8.13 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.58 (d, J = 6.8 Hz, 1H), 7.37 (d, J = 7.2 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.25 (br.s, 1H), 7.18–7.08 (m, 3H), 6.97 (m, 1H), 6.42 (d, J = 7.6 Hz, 1H), 3.36 (m, 4H), 3.31 (m, 4H), 2.33 (s, 3H), 2.26 (s, 3H). 13C-NMR (CDCl3, 100 MHz): δ 157.14, 150.87, 149.55, 148.79, 142.46, 139.37, 136.26, 134.81, 131.76, 129.97, 129.11, 127.88, 127.74, 126.28, 125.56, 125.34, 124.75, 123.77, 123.60, 120.02, 109.01, 52.66, 51.69, 21.22, 20.64; MS (m/z): 425.90 [M + H]+. Anal. Calcd for C27H27N3S: C, 76.20; H, 6.39; N, 9.87; Found: C, 76.47; H, 6.51; N, 9.82.
1-(2-((2,4-dimethylphenyl)thio)phenyl)-4-(5-(trifluoromethyl)pyridin-2-yl) piperazine (27)
Yield: 49%; Off-White Solid. Rf:0.3, AcOEt: Hexane (4:6), 1H-NMR (DMSO-d6, 400 MHz) δ; 8.44 (s, 1H), 7.84 (dd, J = 8.8,2.4 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.24 (br.s, 1H), 7.17–7.09 (m, 3H), 7.04 (d, J = 9.2 Hz, 1H), 6.95–6.91 (m, 1H), 6.42 (d, J = 8.0 Hz, 1H), 3.80 (m, 4H), 3.08 (m, 4H), 2.32 (s, 3H), 2.26 (s, 3H).13C-NMR (CDCl3, 100 MHz) δ; 160.58, 148.78, 145.80, 145.76, 142.39, 139.34, 136.18, 134.65, 134.53, 134.50, 131.75, 127.87, 127.75, 126.34, 125.57, 124.73, 119.81, 114.98, 105.64, 51.43, 45.26, 21.22, 20.63. MS (m/z) 443.97 [M + H]+. Calculated for C24H24F3N3S: C, 64.99; H, 5.45; N, 9.47; Found: C, 64.78; H, 5.29; N, 9.24.
To a solution of vortioxetine (1) (1 eq) in dry DCM (4 mL) solvent, was added N,N-diisopropyl ethylamine(2 eq) at 0 °C, stirred for 5 min, followed by dropwise addition of isocyanate (2 eq). The contents were stirred at room temperature for 2 h and TLC was checked for the completion of reaction. The reaction mixture was quenched using ice water and extracted with DCM (20 mL). The extract was washed with brine, dried over anhydrous Na2SO4, filter and evaporated to get dark colored crude semi-solid material. Crude product was further purified by combiflash column chromatography to get pure solid compound.
N-(3-chloro-4-fluorophenyl)-4-(2-((2,4-dimethylphenyl)thio)phenyl)piperazine-1-carboxamide (28)
Yield: 55%; Light brown solid. Rf:0.3 (AcOEt: Hexane 5:5), 1H-NMR (DMSO-d6, 400 MHz) δ; 8.77 (s, 1H); 7.75 (dd, J = 7.2, 2.8 Hz, 1H), 7.45–7.42 (m, 1H), 7.35–7.24 (m, 3H), 7.17–7.09 (m, 3H), 6.93 (t, J = 8.0 Hz, 1H), 6.42 (d, J = 8.0 Hz, 1H), 3.61 (m, 4H), 3.00 (m, 4H), 2.32 (s, 3H), 2.25 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ; 155.12, 151.56, 149.20, 142.08, 139.61, 138.32, 136.17, 133.87, 132.18, 128.49, 127.70, 126.38, 126.19, 125.10, 121.15, 120.90, 120.02, 119.95, 116.99, 51.67, 44.63, 21.19, 20.58; MS (m/z): 470.10 [M + H]+. Anal. Calcd for C25H25ClFN3OS: C, 63.89; H, 5.36; N, 8.94; Found: C, 64.02; H, 5.39; N, 8.80;
4-(2-((2,4-dimethylphenyl)thio)phenyl)-N-(pyridin-2-yl)piperazine-1-carboxamide (29)
Yield: 50%; Off white solid. Rf:0.35 (AcOEt: Hexane 3:7), 1H-NMR (DMSO-d6, 400 MHz) δ; 8.77 (br s, 1H), 8.66 (d, J = 2.0 Hz,1H), 8.16 (d, J = 3.2 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.29–7.27 (m, 2H), 7.18–7.12 (m, 3H), 6.95 (m, 1H), 6.43 (d, J = 8.0 Hz, 1H), 3.16 (m, 4H), 3.01 (m, 4H), 2.33 (s, 3H), 2.25 (s, 3H). 13C-NMR (CDCl3, 100 MHz,) δ; 155.06, 148.39, 143.63, 142.24, 141.16, 139.28, 136.43, 136.04, 134.54, 131.68, 127.79, 127.68, 126.25, 125.52, 124.78, 123.72, 123.59, 119.85, 51.36, 44.58, 21.11, 20.53; MS (m/z) 419.00 [M + H]+; Anal. Calcd for C24H26N4OS: C, 68.87; H, 6.26; N, 13.39; Found: C, 68.66; H, 6.12; N, 13.54.
Molecular docking
The X-ray crystal structure of SARS-CoV-2 Mpro complexed with co-ligand (N3) PDB ID: 6LU7 (2.16 Å) [6], was retrieved from Research Collaboratory for Structural Bioinformatics RCSB Protein Data Bank (www.rcsb.org). The Mpro crystal structure was imported in AutoDock Tools 1.5.6 [34] and removed water molecules and hetero atoms, and then added polar hydrogen’s followed by computing Gasteiger and adding Kollman charge. Finally, the protein was saved in pdbqt format. The OpenBabel software was used to convert ligands into PDB format [35]. Furthermore, the ligands were prepared by detecting the torsion root, correcting the torsion angles, assigning charges, optimizing using UFF [36] and finally converted into pdbqt format.
In this study, docking was performed using ADV in PyRx virtual screening open-source software [37]. The protein and ligand molecules to be docked are selected under the vina wizard control. The grid was generated by selecting the co-crystallized ligand and grid size can be adjusted according to the active site residues. A grid box with the size 58 × 68 × 70 with coordinates of center_x = -10.883, y = 13.934, and z = 68.209. During docking the grid spacing and exhaustiveness were 0.375 Å and 50, respectively. The docking Lamarckian Genetic Algorithm (LGA) was used [38].
These compounds were again re-docked using AD [39] to eliminate false positive software considering identical receptor grid coordinates. The top docking pose of ADV output file was visualized using Discovery Studio 2020 Client (BIOVIA 2016) software. The virtual screening and ADME were performed using Windows 10 OS in a 64-bit machine, Core 2 Duo CPU microprocessor with 4 GB RAM.
In-silico ADME and drug-likeness prediction
In silico Absorption, Distribution, Metabolism, Elimination (ADME) prediction is one of the important as well as significant criteria to estimate drug-likeness of the selected hits. Conventionally, ADME properties of drug molecules were determined in the last stage of the drug discovery process. In modern drug discovery, ADME properties can be predicted using the in-silico method in the early stage. Due to poor ADME properties, 60% of drug molecules failed in the development process. Therefore, early prediction of these properties would lead to the reduction of drug discovery costs [40]. In the present study, the potential hits were subjected to ADME prediction using a publicly available online web server: SwissADME (http://www.swissadme.ch [41]. Several properties like molecular weight, number of heavy atoms, number of aromatic heavy atoms, number of rotatable bonds, molar refractivity, topological polar surface area, solubility, gastrointestinal absorption, blood–brain, barrier penetration, Lipinski's rule of five, Ghose rule, Veber rule, bioavailability score, and synthetic susceptibility were predicted.
References
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Tao HuT, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74. https://doi.org/10.1016/S0140-6736(20)30251-8.
Gorbalenya AE, Gulyaeva AA, Lauber C, Sidorov IA, Leontovich AM, Penzar D, Samborskiy DV, Baker SC, Baric RS, de Groot RJ, Drosten C, Haagmans BL, Neuman BW, Perlman S, Poon LLM, Sola I, Ziebuhr J. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–44. https://doi.org/10.1038/s41564-020-0695-z.
Das C, Paul SS, Saha A, Singh T, Saha A, Im J, Biswas G. Silver-Based Nanomaterials as Therapeutic Agents Against Coronaviruses: A Review. Int J Nanomed. 2020;15:9301–15. https://doi.org/10.2147/IJN.S280976.
Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden van Noort GJ, Ovaa H, Müller S, Knobeloch KP, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S, Dikic I. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–62. https://doi.org/10.1038/s41586-020-2601-5.
Mody V, Ho J, Wills S, Mawri A, Lawson L, Ebert MCCJC, Fortin GM, Rayalam S, Taval S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun Biol. 2021;4:93. https://doi.org/10.1038/s42003-020-01577-x.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93. https://doi.org/10.1038/s41586-020-2223-y.
Dai W, Zhang B, Jiang X-M, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C, Li Y, Bai F, Wang H, Cheng X, Cen X, Hu S, Yang X, Wang J, Liu X, Xiao G, Jiang H, Rao Z, Zhang L-K, Xu Y, Yang H, Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–5. https://doi.org/10.1126/science.abb4489.
Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–27. https://doi.org/10.1038/s41586-020-2798-3.
Konwar M, Sarma D. Advances in developing small molecule SARS 3CLpro inhibitors as potential remedy for corona virus infection. Tetrahedron. 2021;77: 131761. https://doi.org/10.1016/j.tet.2020.131761.
Tian D, Liu Y, Liang C, Xin L, Xie X, Zhang D, Wan M, Li H, Fu X, Liu H, Cao W. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed Pharmacother. 2021;137:111313. https://doi.org/10.1016/j.biopha.2021.111313.
Paul SS, Biswas G. Repurposed Antiviral Drugs for the Treatment of COVID-19: Syntheses, Mechanism of Infection and Clinical Trials. Mini Rev Med Chem. 2021;21(9):1123–43. https://doi.org/10.2174/1389557521666201222145842.
Deb B, Debnath S, Chakraborty A, Majumdar S. Bis-indolylation of aldehydes and ketones using silica-supported FeCl3: molecular docking studies of bisindoles by targeting SARS-CoV-2 main protease binding sites. RSC Adv. 2021;11:30827–39. https://doi.org/10.1039/D1RA05679D.
Balaramnavar VM, Ahmad K, Saeed M, Ahmad I, Kamal M, Jawed T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 M pro. RSC Adv. 2020;10:40264–75. https://doi.org/10.1039/D0RA06038K.
Lem FF, Opook F, Lee DJH, Chee FT, Lawson FP, Chin SN. Molecular Mechanism of Action of Repurposed Drugs and Traditional Chinese Medicine Used for the Treatment of Patients Infected With COVID-19: A Systematic Scoping Review. Front Pharmacol. 2021;11. https://doi.org/10.3389/fphar.2020.585331
Nguyen HL, Thai NQ, Truong DT, Li MS. Remdesivir Strongly Binds to Both RNA-Dependent RNA Polymerase and Main Protease of SARS-CoV-2: Evidence from Molecular Simulations. J Phys Chem B. 2020;124:11337–48. https://doi.org/10.1021/acs.jpcb.0c07312.
Bolarin JA, Oluwatoyosi MA, Orege JI, Ayeni EA, Ibrahim YA, Adeyemi SB, Tiamiyu BB, Gbadegesin LA, Akinyemi TO, Odoh CK, Umeobi HI, Adeoyej AB-E. Therapeutic drugs for SARS-CoV-2 treatment: Current state and perspective. Int Immunopharmacol. 2021;90:107228. https://doi.org/10.1016/j.intimp.2020.107228.
Ali MJ, Hanif M, Haider MA, Ahmed MU, Sundas F, Hirani A, Khan AI, Anis K, Karim AH. Treatment Options for COVID-19: A Review. Front Med. 2020;7:480. https://doi.org/10.3389/fmed.2020.00480.
Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS, Reiersen AM. Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19. A randomizedclinical trial JAMA. 2020;324:2292–300. https://doi.org/10.1001/jama.2020.22760.
Schloer S, Brunotte L, Mecate-Zambrano A, Zheng S, Tang J, Ludwig S, Rescher U. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-CoV-2 infection in vitro. Br J Pharmacol. 2021;178:2339–50. https://doi.org/10.1111/bph.15418.
Xiao X, Wang C, Chang D, Wang Y, Dong X, Jiao T, Zhao Z, Ren L, Cruz CSD, Sharma L, Lei X, Wang J. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front Immunol. 2020;11:586572. https://doi.org/10.3389/fimmu.2020.586572.
Xiong H-L, Cao J-L, Shen C-G, Ma J, Qiao X-Y, Shi T-S, Ge S-X, Ye H-M, Zhang J, Yuan Q, Zhang T-Y, Xia N-S. Several FDA-Approved Drugs Effectively Inhibit SARS-CoV-2 Infection in vitro. Front Pharmacol. 2021;11: 609592. https://doi.org/10.3389/fphar.2020.609592.
Talmon M, Rossi S, Pastore A, Cattaneo CI, Brunelleschi S, Fresu LG. Vortioxetine exerts anti-inflammatory and immunomodulatory effects on human monocytes/macrophages. Br J Pharmacol. 2018;175:113–24. https://doi.org/10.1111/bph.14074.
Suryavanshi HR, Rathore MM. Synthesis and biological activities of piperazine derivatives as antimicrobial and antifungal agents. Org Commun. 2017;10:228–38. https://doi.org/10.25135/acg.oc.23.17.05.026
Talmon M, Chaudhari RD, Suryavanshi H, Chowdhury N, Quaregna M, Pin A, Bagchi A, Biswas G, Fresu GL. Design, synthesis and biological evaluation of vortioxetine derivatives as new COX-1/2 inhibitors in human monocytes. Bioorg Med Chem. 2020;28: 115760. https://doi.org/10.1016/j.bmc.2020.115760.
Mo X, Li Y, Zhu X, Li X, Zhou H, Bi X, Li J. Vortioxetine Derivatives with Amino acid as Promoiety: Synthesis, Activity, Stability and Preliminary Pharmacokinetic Study. J Pharm Sci. 2021;110:3011–9. https://doi.org/10.1016/j.xphs.2021.04.011.
Moazen-Zadeh E, Bayanati S, Ziafat K, Rezaei F, Mesgarpour B, Akhondzadeh S. Vortioxetine as adjunctive therapy to risperidone for treatment of patients with chronic schizophrenia: A randomised, double-blind, placebo-controlled clinical trial. J Psychopharmacol. 2020;34:506–13. https://doi.org/10.1177/0269881120909416.
Ghosh AK, Brindisi M. Organic Carbamates in Drug Design and Medicinal Chemistry. J Med Chem. 2015;58:2895–940. https://doi.org/10.1021/jm501371s.
Sen D, Bhaumik S, Debnath P, Debnath S. Potentiality of Moringa oleifera against SARS-CoV-2: identified by a rational computer aided drug design method. J Biomol Struct Dyn. 2021;1–18. https://doi.org/10.1080/07391102.2021.1898475
Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci. 2003;100:13190–5. https://doi.org/10.1073/pnas.1835675100.
Yoshino R, Yasuo N, Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci Rep. 2020;10:12493. https://doi.org/10.1038/s41598-020-69337-9.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. https://doi.org/10.1016/s0169-409x(00)00129-0.
Mishra S, Dahima R. In Vitro Adme Studies of TUG-891, A GPR-120 Inhibitor Using Swiss ADME Predictor. J Drug Deliv Ther. 2019;9:366–69. https://doi.org/10.22270/jddt.v9i2-s.2710
Isyaku Y, Uzairu A, Uba S. Computational studies of a series of 2-substituted phenyl-2-oxo-, 2-hydroxyl- and 2-acylloxyethylsulfonamides as potent anti-fungal agents. Heliyon. 2020;6: e03724. https://doi.org/10.1016/j.heliyon.2020.e03724.
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. https://doi.org/10.1186/1758-2946-3-33.
Jaillet L, Artemova S, Redon S. IM-UFF: Extending the universal force field for interactive molecular modeling. J Mol Graph Model. 2017;77:350–62. https://doi.org/10.1016/j.jmgm.2017.08.023.
Leach AR, Shoichet BK, Peishoff CE. Prediction of Protein−Ligand Interactions. Docking and Scoring: Successes and Gaps. J Med Chem. 2006;49:5851–5. https://doi.org/10.1021/jm060999m.
Fuhrmann J, Rurainski A, Lenhof H-P, Neumann D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J Comput Chem. 2010;31:1911–8. https://doi.org/10.1002/jcc.21478.
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. https://doi.org/10.1002/jcc.21256.
Silakari O, Singh PK. Chapter 14-ADMET tools: Prediction and assessment of chemical ADMET properties of NCEs in Concepts Exp Protoc Model Informatics Drug Des. 2021. p. 299–320. https://doi.org/10.1016/B978-0-12-820546-4.00014-3
Daina A, Michielin O, Zoete V. Swiss ADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. https://doi.org/10.1038/srep42717.
Acknowledgements
GB and RC acknowledged West Bengal DST Memo No. 403(Sanc.)/STBT-11012(25)/9/2021-ST SEC for support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All the authors declare no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Suryavanshi, H., Chaudhari, R.D., Patil, V. et al. Design, synthesis and docking study of Vortioxetine derivatives as a SARS-CoV-2 main protease inhibitor. DARU J Pharm Sci 30, 139–152 (2022). https://doi.org/10.1007/s40199-022-00441-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40199-022-00441-z