
Abstract
The identification of a number of families of lipid signal molecules since the 1990s created new therapeutic possibilities for a great number of disorders characterized by chronic inflammation and pain. These lipid autacoids have been explored in a great variety of animal models related to inflammation, pain, (neuro-)protection, and repair. Based on the data from these models, as well as on a number of proof of principle studies in the clinic in indications such as neuropathic pain, a new chapter in medicine is about to begin. We would like to introduce the term “Autacoid Pain Medicine” for this chapter. There are, however, a number of methodological and strategic issues to overcome in this field. One of the roadblocks is related to patent strategies around families of these molecules. As this is not always recognized we will present a number of examples.
Similar content being viewed by others


Introduction
Autacoid medicine is based on a therapeutic intervention in a disease process based on the administration of an autacoid or an autacoid derivative. Autacoids are locally produced modulating factors, influencing the function of nearby cells and/or tissues, which are produced on demand and which subsequently are metabolized in the same cells and/or tissues [1].
The word autacoid stems from the Greek autos (self) and akos (medicinal agent or drug). Sometimes we can find the older terms “secretagogues” or “tissue hormones” for such signaling molecules. Autacoid medicine makes use of the innate mechanisms of defense and repair, which are modulated following administration of an autacoid. In order for such administration to be successful, the autacoid needs to reach the target tissues and build up sufficient concentrations at the site of action, and the choice for a delivery vehicle is an important factor for success. Autacoids used for the treatment of eczema, psoriasis and cornea disorders (such as stromal and Fuchs dystrophy) can reach the target tissues relatively simply via topical formulations. Autacoids for treating chronic inflammations of the kidney, liver, heart and brain are creating some new pharmaceutical development challenges. Treatment via autacoids however offer a number of advantages over the treatment using New Chemical Entities (NCEs). As autacoids are endogenous molecules, metabolic pathways are intrinsic to the tissue. This implies no production and accumulation of toxic metabolites, and a quick resolving of non-active autacoids via innate metabolic processes, with less chance for irreversible or troublesome side effects and no long-term safety issues or idiosyncratic reactions. In this paper selected lipid compounds will be discussed, as in the field of lipid autacoids where most experience has been gathered.
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
The History of Autacoids and the Rise of Autacoid Medicine
Between 1912 and 1960, the meaning of the word autacoid was quite different from the current meaning; the word referred to hormone stimulation. Autacoid in its present meaning was used from the 1960s and the first autacoids identified were histamine, serotonin and bradykinin. Bradykinin at that time (1963) was defined as a so-called tissue-hormone, in the sense that there is no specialized gland to secrete it [2]. Serotonin was defined in 1970 per exlusionam as: “Serotonin belongs to the group of autacoids, i.e. substances of intense pharmacological activity that are normally present in the body and that cannot be classed with hormones or neurohumors.” [3, p. 848] Clearly at that time not all requirements for the classification of a compound as an autacoid were transparent. For hormones, neurotransmitters and autacoids, operational definitions had to emerge step by step. In order to define whether a certain substance can be defined as a hormone, a neurotransmitter or an autacoid, a number of criteria need to be fulfilled (see Table 1). Some autacoids can also be classified in another context as neurotransmitters, for instance NO, ATP, and a number of endocannabinoids such as 2-Arachidonoylglycerol (2-AG); certain neurotransmitters can also act as hormones, such as the catecholamines. Most autacoids play an important role in chronic pain states.
The identification of lipid autacoids gave rise to a number of patents and stimulated research in the field of chronic inflammation. However, the use of the term “autacoid” has been in decline since 1992. During that year more than 8000 publications could be identified in PubMed, dropping to around 4000 in 2015 (Fig. 1).

Profile of PubMed search of the term "autacoid". The first publications appeared in 1934, peaked around 1990–1992, and since 1993 there has been a steady decline
The first glimpse of the potential value of autacoid medicine was identified in 1964 focusing on neuroprotection. It was stated: “the value of physiological compounds, added in excess of their normal concentrations, to forestall the first irreversible changes in the cells” [4]. The authors were quite visionary and published the first paper in literature suggesting to explore physiological acting compounds as drugs for cell and tissue protection.
In 1981, Melmom and co-workers pointed out that autacoids should be regarded as moderators of a number of immune functions, and that concentrations of autacoids in tissues during inflammation were sufficient to allow them to modify the functions of a number of cells which control and express both humoral- and cell-mediated immunity [5]. These autacoids were subsequently characterized by them as feedback modifiers connecting the early and late phases of the immune response. They identified the first building block of Autacoid Medicine, when they stated: “We should probably begin devising a strategy for using these “self-medications” as alternatives to drugs that are less selective in their actions.” (pp. 100–101).
They also pointed out the fact that autacoids can influence the immune process at different stages, and that they can influence different subpopulations of cells at concentrations which probably exist endogenously during physiologic and pathologic events, indicates that they can and should be seriously considered not only as ordinary modulators of the inflammatory and immune processes, but also as potential drugs that can ameliorate these pathologic conditions (p. 104).
In 1986, the eminent neuroscientist Professor Erminio Costa (1924–2009) stipulated this principle in an unpublished keynote lecture he delivered in Washington [6]. He emphasized Pharma should learn from nature how to design a pharmacology of modulation. Costa thus pointed out how nature itself can become our tutor in developing new therapeutic inroads, in so far as we can trigger and activate endogenous repair and defense mechanisms of the body by administrating the endogenous molecules related to these pathways. These statements are consistent with the vision of Melmom et al. [5].
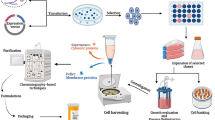
Professor Bruni from the Department of Pharmacology, University of Padova and working for the Italian company Fidia, emphasized that due to the potency and efficacy, autacoids stimulated drug discovery. He was also probably the first author to point out the role of plasma membrane in the production of lipid autacoids [7] (Fig. 2).

Adapted from Autocoids from membrane phospholipids, Bruni A. 1988, Pharmacological Research Communications
Professor Bruni correctly stated in 1988 that one of the major metabolic route for many lipid autacoids is via the plasma membrane
An important family of lipid autacoids was identified by the Nobel laureate Rita Levi-Montalcini, who first described palmitoylethanolamide (PEA) in 1993 and tested a number of derivatives as anti-inflammatory and anti-pain compounds in a series of preclinical models. For these compounds she introduced ALIAmides, as an acronym for ALIA: Autacoid Local Inflammatory Antagonism. A few years later in 1996, the term was refined by Levi-Montalcini in a broader way [8]. She also included the (neuro-) repair function of autacoids, and ALIA was presented as the abbreviation of Autacoid Local Injury Antagonism. In a paper on the protective effects of PEA in central neuron death, she explained: “Palmitoylethanolamide, in reality, appears to exert a more broad local autacoid anti-injury function, thus the acronym autacoid local injury antagonism for ALIA.” Here we see that it was actually Levi-Montalcini anticipated the neuroprotective function of these lipid autacoids.
Classes of Autacoids
There are many different families of autacoids and we suggest six classes, as depicted in Table 2.
The lipid autacoids vary from small signaling lipids to high-molecular-weight glycerophospholipids, with many structural isomers making the profiling of the lipidomic landscape a difficult task. Serhan and his colleagues identified new families of lipids, the lipoxins in the 1980s and the resolvins in resolving inflammatory exudates, (resolution–phase interaction products) in 2002 [9]. The lipid autacoids from the family of “specialized pro-resolving mediators” (SPMs) such as resolvins, protectins, and maresins, are all synthesized via oxidation of omega-3 essential fatty acids precursors. Lipoxin, derived from arachidonic acid, is considered an SPMs also. All the compounds and derivatives from the class SPMs counteract inflammation in tissues triggered by pro-inflammatory prostaglandins and leukotrienes. This holds also true for the lipid autacoids belonging to the N-acylethanolamides such as palmitoylethanolamide, which inhibits such pro-inflammatory signals via the activation of the peroxisome proliferator-activated receptors (PPARs).
After 1992 a number of patents were filed on lipid autacoids, which created a proprietary platform but also seemed to obstruct developmental progress in this field. We will review these patents and patent strategies and discuss some of their shortcomings from the perspective of the pain clinician and their patients.
Patents on Lipid Autacoids and Their Derivatives: Pitfalls of IP Protection for Development
The essence of a patent is to protect an innovative and new idea for the inventor, and the innovation described in a patent is meant to be developed into an application which will serve mankind (IP—Intellectual Property). Patents in the therapeutic field should, therefore, lead to therapies. If patents do not lead to such therapies, there are flaws in the process leading from idea to application. We need to analyze this process in more detail because in autacoid medicine such flaws seem to exist and impair development.
First patents on recognized lipid autacoids were the patents filed by a small Italian company, LifeGroup Spa, a company run in the 1990s by the previous CEO of Fidia, Dr. F. Della Valle. It was in Fidia where much of the lipid autacoid R&D started, as outlined above via the work of Professor Bruni. Della Valle was also a key player in the field (and intimately connected to Erminio Costa and Rita Levi-Montalcini) who organized an intense network of research activity between the academia and his pharmaceutical industry. This served as an example of how to create positive synergies [10, 11]. In the patents from LifeGroup we find results from the research of Levi-Montalcini on PEA and derivatives in mast cell models. At that time PEA was referred to by LifeGroup under the development code LG2110/1 and the priority date of the first patent was 31 December 1991 [12]. The patent described a number of N-acyl-derivatives of hydroxyamines “suitable for the therapeutic treatment of pathologies characterized by degranulation of mast cells caused by a neurogenic and/or immunogenic hyperstimulation”. The one figure in the patent depicted the effects of the oral treatment of PEA as LG2110/1) in a dose of 10 mg/kg BW in an experimental allergic encephalomyelitis (encephalitis) model. This dose had clear positive effects on the neurological outcome. In this patent the synthesis of many N-acyl-derivatives of hydroxyamines were described and a number tested positively in animal models for various inflammatory indications, such as arthritis. Apart from PEA, which has been a known registered drug for the treatment of respiratory viral indications (brand name Palmidrol) since the 1970s, none of the derivatives were ever further developed by LifeGroup or any other pharmaceutical company for any other indication [13]. The patent, therefore, functioned as a road block for other organizations to explore the anti-inflammatory effects and the clinical value of this entire family of compounds. In 1996, LifeGroup Spa’s burn rate was such that further steps to develop PEA and related compound became impossible. The expectation was that PEA would be registered for neuropathic pain (sciatic pain) and possibly for multiple sclerosis within a short timeframe, but these expectations were not met in real life as the Italian authorities took too much time reviewing the application, resulting in the premature end of the company. The lesson learned from this case is perhaps that an extensive Intellectual Proprietary Platform around a family of lipid autacoids needs an aggressive license-out policy in order to disseminate the principle broadly. This did not happen, but happily efforts lead to the market entry of PEA many years later (around 2007), although none of the possibly more promising derivatives were ever developed. For some years patent protection for these compounds stopped, and there is no longer an incentive for any industry to test those PEA derivatives. The same strategic problem has happened with the patent portfolio based on Serhan’s work around resolvins and lipoxins.
Charles N. Serhan and his hospital filed a great number patents to protect many inventions related to a number of lipid autacoid families such as the lipoxins, resolvins, maresins, protectins, docosatrienes, and their analogues [14]. The first patent on lipoxin A and B was filed in 1984 (priority date May 4, 1984) [15]. Many more patents in this field have been filed since by Serhan et al. starting at the end of the last century [16,17,18,19,20,21]. However, since May 1, 2017, over 1600 publications on lipoxins can be found in PubMed, but only one clinical pilot trial in atopic eczema can be identified.
In one of the first patents from Serhan in 1994, a number of putative indications are summarized for new lipoxin derivatives as having superior pharmacokinetic properties compared to lipoxin A and B: “Compounds having the active site of natural lipoxins, but a longer tissue half-life are disclosed. These small molecules are useful for treating vasoconstrictive, inflammatory, myeloid suppressive, cardiovascular, and gastrointestinal diseases” [22]. Lipoxins at that time were already known as antagonists to leukotrienes, important mediators of inflammation. The patent claims compounds with the same bioactivities as natural lipoxins, but characterized by a longer in vivo half-life time due to a new metabolic transformation region in the molecule of either lipoxin A4 or B4, resistant to in vivo metabolism. Moreover, the long series of derivatives claimed in the patent (many hundreds of molecules) were synthesized to be more lipophilic than the natural lipoxins in order to enhance their uptake via biological membranes. A number of claims (10–20) in this patent were related to these lipoxin derivatives to be used as drugs in the treatment of inflammation or an inflammatory response in rheumatoid arthritis or induced by physical trauma and radiation exposure. In addition, a great number of potential indications were summarized, some related to pain states: anaphylactic reactions, allergic reactions, shock, inflammation, gout, psoriasis, allergic rhinitis, adult respiratory distress syndrome, Crohn’s disease, endotoxin shock, traumatic shock, hemorrhagic shock, bowel ischemic shock, renal glomerular disease, benign prostatic hypertrophy, inflammatory bowel disease, myocardial ischemia, myocardial infarction, circulatory shock, brain injury, systemic lupus erythematosus, glomerular kidney disease and myeloid suppressive disorders. Not long afterwards, lipoxins were demonstrated to have impressive effects as TNF-alpha inhibitors, and thus the potential field of neuropathic pain opened [23].
None of the derivatives of the lead compound lipoxin A4 have been taken up in clinical development since 1994 [24]. In 2015 in a review on lipoxins we found the statement: “Since lipoxins … have clinical relevance, we discuss their important role in clinical research to treat a wide range of diseases like inflammatory disorders, renal fibrosis, cerebral ischemia, and cancer” [25]. Although much research has been conducted in this field, this has led to 1599 preclinical publications and only one reported clinical pilot trial. It seems that the many lipoxin-related patents filed since 1984, summarized above, did not substantiate into even one clinical available drug.
For patents, it seems that a sunset clause is missing: this would be a statutory provision providing that a particular patent will expire on a particular date, unless it is reauthorized by the legislature based on clear and explicate activities related to its research and development. This would at least stimulate the patent holders to translate the patent findings into hands on development activities, or to license a patent in an earlier phase to third parties.
Putative Indications of Lipid Autacoids Related to Chronic Inflammation and Pain
The relevance of lipid autacoids for the treatment of chronic disorders is vast, and especially since many lipid autacoids possess clear pro-resolving and anti-inflammatory activities the putative indications are many. We point out just a few and for the sake of argumentation we select for each class the same indications.
Resolvins
Alzheimer’s disease [26], depression [27], sepsis [28], asthma [29], reperfusion injury [30, 31], glaucoma and retinitis [32], obesitas [33], auto-immune inflammatory demyelination disorders [34], arthritis [35], osteoarthritis [36], infections [37], sciatic [38] and neuropathic pain [39].
Endocannabinoids
Alzheimer’s disease [40], depression [41], sepsis [42], asthma [43], reperfusion injury [44], glaucoma and retinitis [45], obesitas [46], auto-immune inflammatory demyelination disorders [47], arthritis [48], osteoarthritis [49], infections [50], sciatic [51] and neuropathic pain [52].
N-Acylethanolamides
In most indications explored the N-acylethanolamide palmitoylethanolamide has been tested. This is related to the fact that PEA has been available in a number of European countries since 2007 as a nutraceutical.
Palmitoylethanolamide: Alzheimer’s disease [53], depression [54], sepsis [55], asthma [56], reperfusion injury [57], glaucoma and retinitis [58], obesitas [59], auto-immune inflammatory demyelination disorders [60], arthritis [61], osteoarthritis [62], infections [63], sciatic [64] and neuropathic pain [65].
In the list above it is clear that there is a major overlap and representatives from each class of lipids are evaluated positively in the respective models for these disorders. In most of these disorders the animal models have generated supportive data, and in some cases positive clinical trials can be seen as proof of principle. Such clinical trials support the use of certain lipid autacoids such as PEA in neuropathic pain. Of course, we need to mention that positive clinical studies also support or suggest the use of Cannabis in the same list of disorders, including as a key treatment of various pain states. Mechanisms of actions of Cannabis, endocannabinoids and lipid signaling molecules belonging to the autacoids clearly overlap.
Lipid Autacoids as Neuroprotectants and Analgesics in Models of Neuropathic Pain
Lipid autacoids have been explored in a great number of in vitro and in vivo test models related to the disorders of the central and peripheral nervous system. In this section we will discuss the analgesic efficacy of RvE1 and PEA in models for peripheral neuropathic pain. However, RvE1 and its derivatives are not yet available for the clinician and PEA in various formulations has been available in Europe since 2007 as a nutraceutical so prescriptions are not required.
Resolvins
Resolvins for the treatment of neuropathic pain have been claimed in a patent of 2009 [66]. In this patent a number of resolvins (hundreds, as well as salts and esters) were described, active in the nanogram dose range, producing anti-hyperalgesic effects in models for inflammatory and neuropathic pain. Many of these findings were presented in a paper in 2010 [67]. RvE1 was reported to reduce pain in mice models for central and peripheral pain after (intrathecal) administration of very a low dose: 0.3 and 1.0 and 10 ng, and was more effective compared to 10 μg of the COX-2 inhibitor NS-398 [67]. A stable metabolic analogue 19-(p-fluorophenoxy)-RvE1 (19-pf-RvE1) had a comparable, but longer effect after intrathecal administration: 6 h. Peripheral administered RvE1 also led to reduced intraplantar capsaicin-induced acute pain. However, all administration of the resolvin and its analogues was parenteral and led to relatively short-lived effects. The experiments led the authors to state that resolvins and their metabolically stable analogs may represent a new family of analgesics useful in treating inflammation-associated pain states such as arthritic and post-operative pain. This position was also taken in the review in Nature Medicine in 2010, where the above-mentioned findings were discussed and where again it was pointed out that these results seem to be very promising for the development of a drug to be used in humans [67]. This argument was based on only one element of the compounds tested, the nanomolar and picomolar potency, which was regarded as a desirable characteristic for any candidate drug. The challenge, the authors concluded, would be to develop resolvins into a clinically applicable form.
RvE1 is presented as a new therapeutic target for the treatment of inflammatory pain as it strongly inhibits TRPV1 activity associated with peripheral inflammation [68]. In addition, there are central mechanisms of action, on glia level. Such findings support researchers to express the belief, given the potent analgesic efficacy and safety profiles of endogenous lipid mediators that resolvins, their metabolically stable analogues and small molecule agonists that target the resolvin’s signaling pathways, may offer new therapeutic tools for the management of neuropathic pain [69]. Also, pathomechanistic studies spinal (intrathecal) pretreatment of RvE1 (100 ng) in a chronic constriction injury of the sciatic nerve (CCI) could reduce the mechanical allodynia 24 h after administration [70].
Palmitoylethanolamide
With over 600 PubMed indexed papers on palmitoylethanolamide (PEA), this lipid autacoid is by far the best explored in preclinical, as well as in clinical studies.
All studies are in line with its analgesic and anti-inflammatory actions in a dose range of 10–30 mg/kg body weight. Doses up to 100 mg/kg body weight have been administered to humans without any evidence of adverse effects. Given its very benign safety profile, the compound is also fit for elderly and multi-morbid patients [71]. For the clinical data on PEA, we refer to the many review articles written since 2012. PEA seems safe and effective in a range of neuropathic pain syndromes, from painful diabetic neuropathy to pain in chronic idiopathic axonal neuropathy [72,73,74,75]. In nerve compression syndromes, such as sciatic pain and carpal tunnel syndrome, a series of placebo controlled studies support the safe and effective use of PEA in these indications. Analysis of data from an RCT in over 600 sciatic pain patients demonstrated a NNT for PEA of 1.5 [76]. Its neuroprotective profile has been evaluated in the ophthalmological clinic where the compound proved to possess retinoprotective properties [77].
The author has worked with PEA in his clinic for neuropathic pain since 2010 and has treated many thousands of patients, mainly those with axonal neuropathies such as diabetic neuropathy and chronic idiopathic axonal neuropathy (CIAP) and sciatic pain [78]. Based on his experience and results of recent RCTs, he recommends a dose of 1200 mg daily in neuropathic pain, for at least 2 months. This time frame seems to help the autacoid to reset the system. He also found that partial responders sometimes became full responders after doubling this dose. He has used a number of formulations of PEA, and he and other Dutch colleagues found only patented formulations containing microPEA formulations to be effective and safe. Patients have indicated that use of cheap “me-toos” lead to suboptimal efficacy, most probably due to the absence of an optimal formulation. There are only three micro-PEA formulations based on patent protected know-how of formulations of PEA with adequate bioavailability: PEA-um (ultramicronized), PEA-m (micronized), and PEA-opt (optimized). For a crystalline lipid as PEA, formulations containing micro-PEA seem key for the clinical success. Studies supporting the safety and efficacy of other PEA formulations to date are absent.
Conclusion
Lipid autacoids and their derivatives move tissue towards balance in the case of overactive inflammation, and this is one of the key elements in neuropathic pain. The various classes of lipid autacoids such as the lipoxins, protectins, resolvins, N-acylethanolamides, and endocannabinoids have been extensively tested positively in animal models for a great variety of disorders, all characterized by (neuro-)inflammation, including in pain models. PEA is the most advanced compound, as this molecule has been extensively tested since the 1970s and is available as a nutraceutical without the necessity of a prescription. This is a both a strength and a weakness; for patients the compound is easily accessible, for pain physicians the compound is outside of their (pharma-)scope.
Research in this field of Autacoid Medicine has been hampered by the many patents filed in the 1990s, and the absence of serious drug development in this field because the majority of research was driven by academia. Many compounds are mentioned in these patents, compounds with great promise, which have never been tested nor licensed out by the inventors. Formulation development in this field is also rare, and as most compounds have a short half-life time, such work is mandatory for its success.
Lipid autacoids hold great hope for pain medicine especially since its side-effect profiles will be benign, and as the metabolic routes are endogenous no surprises are to be expected related to their metabolites. Now that a great number of patents have expired, new possibilities for formulation of used patents may re-open the path to serious Research and Development.
References
Keppel Hesselink JM. The terms ‘autacoid’, ‘hormone’ and ‘chalone’ and how they have shifted with time. Auton Autacoid Pharmacol. 2015;35:51–8.
Silva M. The physiological significance of bradykinin. Ann N Y Acad Sci. 1963;104(1):190–211.
Sirek A, Sirek OV. Serotonin: a review. Can Med Assoc J. 1970;102(8):846.
Lewis Wright R, Ames Adelbert III. Measurement of maximal permissible cerebral ischemia and a study of its pharmacologic prolongation*. J Neurosurg. 1964;21(7):567–74.
Melmon KL, Rocklin RE, Rosenkranz RP. Autacoids as modulators of the inflammatory and immune response. Am J Med. 1981;71(1):100–6.
Costa E. To follow where nature leads. Washington: Lectio Magistralis; 1986.
Bruni A. Autacoids from membrane phospholipids. Pharmacol Res Commun. 1988;20(7):529–44.
Levi-Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19(11):514–20.
Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac R-L. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. J Exp Med. 2002;196:1025–37.
Della Valle F. FIDIA-Georgetown Institute for the Neurosciences: dreams, projects and actions with Erminio Costa. Pharmacol Res. 2011;64(4):310–1.
Keppel Hesselink JM. Palmitoylethanolamide: research Synergy between academia and industry, based on insights and work of nobel laureate rita levi-montalcini. J Clin Trials Pat. 2016;1(1):3.
Della Valle F., Silvana Lorenzi, Jacobus C. J. J. Samson, and Federica della Valle. N-acyl derivatives of aminoalcohols active as local autacoids and useful in the therapy of autoimmune processes. Patent US 5506224 A, priority date 31 December 1991.
Keppel Hesselink JM, de Boer T, Witkamp RF. Palmitoylethanolamide: a natural body-own anti-inflammatory agent, effective and safe against influenza and common cold. Int J Inflam. 2013;2013:151028.
Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA. 1984;81(17):5335–9.
Samuelsson B, Serhan C, and Hamberg M. Inflammatory lipoxin A and anti-inflammatory lipoxin B compounds US 4560514 A, priority date 4 May 1984.
Serhan CN, and Badr K. Use of lipoxin a4 and its derivatives as antagonists for slow-reacting substances of anaphylaxis, WO1990013292A1, priority date 27 April 1989.
Serhan CN. Lipoxin compounds, WO 1994029262 A1, priority date 15 June 1994).
Serhan CN. Lipoxin compounds and their use in treating cell proliferative disorders, WO1998011049A1, priority date 15 June 1994.
Serhan CN. Inhibition of TNF-alpha-initiated neutrophil response, US 20040192785 A1, priority date 18 March 1999.
Serhan CN. Aspirin-triggered lipid mediators, WO2001060778A2, priority date 16 February 2000.
Van Dyke TE, Petasis NA, and Serhan CN. Lipoxin analogs and methods for the treatment of periodontal disease, WO2001070664A2, priority date 20 March 2000.
Serhan CN. Lipoxin compounds, US 5441951 A, priority date 15 June 1994.
Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J Exp Med. 1999;189(12):1923–30.
Wu SH, Chen XQ, Liu B, Wu HJ, Dong L. Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br J Dermatol. 2013;168(1):172–8.
Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature’s way to resolve inflammation. J Inflamm Res. 2015;30(8):181–92.
Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC, Serhan CN, Schultzberg M. Pro-resolving lipid mediators improve neuronal survival and increase Aβ42 phagocytosis. Mol Neurobiol. 2016;53(4):2733–49.
Klein CP, Sperotto ND, Maciel IS, Leite CE, Souza AH, Campos MM. Effects of d-series resolvins on behavioral and neurochemical changes in a fibromyalgia-like model in mice. Neuropharmacology. 2014;86:57–66.
Dalli J, Colas RA, Quintana C, Barragan-Bradford D, Hurwitz S, Levy BD, Choi AM, Serhan CN, Baron RM. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: correlations with survival and clinical outcomes. Crit Care Med. 2016;45(1):58–68.
Zambalde ÉP, Teixeira MM, Favarin DC, de Oliveira JR, Magalhães ML, Cunha MM, Silva WC, Okuma CH, Rodrigues V, Levy BD, Rogerio AP. The anti-inflammatory and pro-resolution effects of aspirin-triggered RvD1 (AT-RvD1) on peripheral blood mononuclear cells from patients with severe asthma. Int Immunopharmacol. 2016;35:142–8.
Kang JW, Lee SM. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim Biophys Acta. 2016;1861(9):1025–35.
Zhao Q, Wu J, Lin Z, Hua Q, Zhang W, Ye L, Wu G, Du J, Xia J, Chu M, Hu X. Resolvin D1 alleviates the lung ischemia reperfusion injury via complement, immunoglobulin, TLR4, and inflammatory factors in rats. Inflammation. 2016;39(4):1319–33. doi:10.1007/s10753-016-0364-9.
Keppel Hesselink JM, Chiosi F, Costagliola C. Resolvins and aliamides: lipid autacoids in ophthalmology—what promise do they hold? Drug Des Devel Ther. 2016;10:3133–41.
Titos E, Rius B, López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Lopategi A, Dalli J, Lozano JJ, Arroyo V, Delgado S, Serhan CN, Clària J. Signaling and immunoresolving actions of resolvin D1 in inflamed human visceral adipose tissue. J Immunol. 2016;197(8):3360–70.
Luo B, Han F, Xu K, Wang J, Liu Z, Shen Z, Li J, Liu Y, Jiang M, Zhang ZY, Zhang Z. Resolvin D1 programs inflammation resolution by increasing TGF-β expression induced by dying cell clearance in experimental autoimmune neuritis. J Neurosci. 2016;36(37):9590–603.
Arnardottir HH, Dalli J, Norling LV, Colas RA, Perretti M, Serhan CN. Resolvin D3 is dysregulated in arthritis and reduces arthritic inflammation. J Immunol. 2016;197(6):2362–8.
Benabdoune H, Rondon EP, Shi Q, Fernandes J, Ranger R, Fahmi H, Benderdour M. The role of resolvin D1 in the regulation of inflammatory and catabolic mediators in osteoarthritis. Inflamm Res. 2016;65(8):635–45.
Norris PC, Arnardottir H, Sanger JM, Fichtner D, Keyes GS, Serhan CN. Resolvin D3 multi-level proresolving actions are host protective during infection. Prostaglandins Leukot Essent Fatty Acids. 2016;. doi:10.1016/j.plefa.2016.01.001.
Liu ZH, Miao GS, Wang JN, Yang CX, Fu ZJ, Sun T. Resolvin D1 Inhibits Mechanical Hypersensitivity in Sciatica by Modulating the Expression of Nuclear Factor-κB, Phospho-extracellular Signal-regulated Kinase, and Pro- and Anti-inflammatory Cytokines in the Spinal Cord and Dorsal Root Ganglion. Anesthesiology. 2016;124(4):934–44.
Xu ZZ, Berta T, Ji RR. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J Neuroimmune Pharmacol. 2013;8(1):37–41.
Fernández-Ruiz J, Romero J, Ramos JA. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s Chorea, Alzheimer’s disease, and others. Handb Exp Pharmacol. 2015;231:233–59.
Gorzalka BB, Hill MN. Putative role of endocannabinoid signaling in the etiology of depression and actions of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1575–85. doi:10.1016/j.pnpbp.2010.11.021.
Kianian M, Al-Banna NA, Kelly ME, Lehmann C. Inhibition of endocannabinoid degradation in experimental endotoxemia reduces leukocyte adhesion and improves capillary perfusion in the gut. J Basic Clin Physiol Pharmacol. 2013;24(1):27–33.
Zoerner AA, Stichtenoth DO, Engeli S, Batkai S, Winkler C, Schaumann F, Janke J, Holz O, Krug N, Tsikas D, Jordan J, Hohlfeld JM. Allergen challenge increases anandamide in bronchoalveolar fluid of patients with allergic asthma. Clin Pharmacol Ther. 2011;90(3):388–91.
Pacher P, Haskó G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2008;153(2):252–62.
Chen J, Matias I, Dinh T, Lu T, Venezia S, Nieves A, Woodward DF, Di Marzo V. Finding of endocannabinoids in human eye tissues: implications for glaucoma. Biochem Biophys Res Commun. 2005;330(4):1062–7.
Kim J, Li Y, Watkins BA. Fat to treat fat: emerging relationship between dietary PUFA, endocannabinoids, and obesity. Prostaglandins Other Lipid Mediat. 2013;104–105:32–41.
Pryce G, Baker D. Endocannabinoids in Multiple Sclerosis and Amyotrophic Lateral Sclerosis. Handb Exp Pharmacol. 2015;231:213–31.
Gui H, Tong Q, Qu W, Mao CM, Dai SM. The endocannabinoid system and its therapeutic implications in rheumatoid arthritis. Int Immunopharmacol. 2015;26(1):86–91.
La Porta C, Bura SA, Llorente-Onaindia J, Pastor A, Navarrete F, García-Gutiérrez MS, De la Torre R, Manzanares J, Monfort J, Maldonado R. Role of the endocannabinoid system in the emotional manifestations of osteoarthritis pain. Pain. 2015;156(10):2001–12.
Patsenker E, Sachse P, Chicca A, Gachet MS, Schneider V, Mattsson J, Lanz C, Worni M, de Gottardi A, Semmo M, Hampe J, Schafmayer C, Brenneisen R, Gertsch J, Stickel F, Semmo N. Elevated levels of endocannabinoids in chronic hepatitis C may modulate cellular immune response and hepatic stellate cell activation. Int J Mol Sci. 2015;16(4):7057–76.
Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330(3):902–10.
Maldonado R, Baños JE, Cabañero D. The endocannabinoid system and neuropathic pain. Pain. 2016;157(Suppl 1):S23–32.
Cipriano M, Esposito G, Negro L, Capoccia E, Sarnelli G, Scuderi C, De Filippis D, Steardo L, Iuvone T. Palmitoylethanolamide regulates production of pro-angiogenic mediators in a model of β amyloid-induced astrogliosis in vitro. CNS Neurol Disord Drug Targets. 2015;14(7):828–37.
Yu HL, Deng XQ, Li YJ, Li YC, Quan ZS, Sun XY. N-palmitoylethanolamide, an endocannabinoid, exhibits antidepressant effects in the forced swim test and the tail suspension test in mice. Pharmacol Rep. 2011;63(3):834–9.
Redlich S, Ribes S, Schütze S, Nau R. Palmitoylethanolamide stimulates phagocytosis of Escherichia coli K1 by macrophages and increases the resistance of mice against infections. J Neuroinflammation. 2014;14(11):108.
Yoshihara S, Morimoto H, Ohori M, Yamada Y, Abe T, Arisaka O. Endogenous cannabinoid receptor agonists inhibit neurogenic inflammations in guinea pig airways. Int Arch Allergy Immunol. 2005;138(1):80–7.
Impellizzeri D, Bruschetta G, Ahmad A, Crupi R, Siracusa R, Di Paola R, Paterniti I, Prosdocimi M, Esposito E, Cuzzocrea S. Effects of palmitoylethanolamide and silymarin combination treatment in an animal model of kidney ischemia and reperfusion. Eur J Pharmacol. 2015;5(762):136–49.
Keppel Hesselink JM, Costagliola C, Fakhry J, Kopsky DJ. Palmitoylethanolamide, a natural retinoprotectant: its putative relevance for the treatment of glaucoma and diabetic retinopathy. J Ophthalmol. 2015;2015:430596.
Mattace Raso G, Santoro A, Russo R, Simeoli R, Paciello O, Di Carlo C, Diano S, Calignano A, Meli R. Palmitoylethanolamide prevents metabolic alterations and restores leptin sensitivity in ovariectomized rats. Endocrinology. 2014;155(4):1291–301.
Loría F, Petrosino S, Mestre L, Spagnolo A, Correa F, Hernangómez M, Guaza C, Di Marzo V, Docagne F. Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur J Neurosci. 2008;28(4):633–41.
Bonezzi FT, Sasso O, Pontis S, Realini N, Romeo E, Ponzano S, Nuzzi A, Fiasella A, Bertozzi F, Piomelli D. An important role for N-acylethanolamine acid amidase in the complete Freund’s adjuvant rat model of arthritis. J Pharmacol Exp Ther. 2016;356(3):656–63.
Marini I, Bartolucci ML, Bortolotti F, Gatto MR, Bonetti GA. Palmitoylethanolamide versus a nonsteroidal anti-inflammatory drug in the treatment of temporomandibular joint inflammatory pain. J Orofac Pain. 2012;26(2):99–104.
Rasková H, Masek K, Linèt O. Non-specific resistance induced by palmitoylethanolamide. Toxicon. 1972;10(5):485–90.
KeppelHesselink JM, Kopsky DJ. Palmitoylethanolamide, a nutraceutical, in nerve compression syndromes: efficacy and safety in sciatic pain and carpal tunnel syndrome. J Pain Res. 2015;8:729–34.
Keppel Hesselink JM. Chronic idiopathic axonal neuropathy and pain, treated with the endogenous lipid mediator palmitoylethanolamide: a case collection. Int Med Case Rep J. 2013;13(6):49–53.
Ji RR, and Serhan CN. A method for treating neuropathic pain, WO 2011034887 A2, priority date September 15, 2009.
Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, Serhan CN, Ji R. Resolvins RvE1 and RvD1attenuate inflammatory pain via central and peripheral actions. Nat Med. 2010;16(5):592–7.
Jo YY, Lee JY, Park CK. Resolvin E1 inhibits substance P-induced potentiation of TRPV1 in primary sensory neurons. Mediators Inflamm. 2016;2016:5259321.
Xu ZZ, Berta T, Ji RR. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J Neuroimmune Pharmacol. 2013;8(1):37–41.
Xu ZZ, Berta T. Ji R. R. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J Neuroimmune Pharmacol. 2013;8(1):37–41.
Paladini A, Fusco M, Coaccioli S, Skaper SD, Varrassi G. Chronic pain in the elderly: the case for new therapeutic strategies. Pain Physician. 2015;18(5):E863–76.
Keppel Hesselink JM. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. Open Pain J. 2012;5:12–23.
Keppel Hesselink JM, Hekker TA. Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: a case series. J Pain Res. 2012;5:437–42.
Keppel Hesselink JM. Chronic idiopathic axonal neuropathy and pain, treated with the endogenous lipid mediator palmitoylethanolamide: a case collection. Int Med Case Rep J. 2013;13(6):49–53.
Keppel Hesselink JM. Pain management. In: Flint G, Rusbridge C, editors. Syringomyelia, A Disorder of CSF Circulation. Berlin: Springer-Verlag; 2014. p. 237–60.
Keppel Hesselink JM, Kopsky DJ. Palmitoylethanolamide, a nutraceutical, in nerve compression syndromes: efficacy and safety in sciatic pain and carpal tunnel syndrome. J Pain Res. 2015;8:729–34.
Keppel Hesselink JM, Costagliola C, Fakhry J, Kopsky DJ. Palmitoylethanolamide, a natural retinoprotectant: its putative relevance for the treatment of glaucoma and diabetic retinopathy. J Ophthalmol. 2015;. doi:10.1155/2015/430596.
Keppel Hesselink JM. Chronic idiopathic axonal neuropathy and pain, treated with the endogenous lipid mediator palmitoylethanolamide: a case collection. Int Med Case Rep J. 2013;13(6):49–53.
Acknowledgements
No funding or sponsorship was received for this study or the publication of this article. The named author meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, takes responsibility for the integrity of the work as a whole, and has given final approval for the version to be published.
Disclosures
Jan M. Keppel Hesselink has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Content
To view enhanced content for this article go to http://www.medengine.com/Redeem/1A98F0600039E3E6.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keppel Hesselink, J.M. Fundamentals of and Critical Issues in Lipid Autacoid Medicine: A Review. Pain Ther 6, 153–164 (2017). https://doi.org/10.1007/s40122-017-0075-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-017-0075-4