
Abstract
Introduction
Asunaprevir (ASV) is a potent, pangenotypic, twice-daily hepatitis C virus (HCV) NS3 inhibitor indicated for the treatment of chronic HCV infection.
Methods
A population pharmacokinetic (PPK) model was developed using pooled ASV concentration data from 1239 HCV-infected subjects who received ASV either as part of the DUAL regimen with daclatasvir or as part of the QUAD regimen with daclatasvir and peg-interferon/ribavirin.
Results
A two-compartment model with first-order elimination from the central compartment, an induction effect on clearance, and an absorption model consisted of zero-order release followed by first-order absorption adequately described ASV PK after oral administration. A typical value for ASV clearance (CL/F) was 50.8 L/h, increasing by 43% after 2 days to a CL/F of 72.5 L/h at steady-state, likely due to auto-induction of cytochrome P450 3A4 (CYP3A4). Factors indicative of hepatic function were identified as key influential covariates on ASV exposures. Subjects with cirrhosis had an 84% increase in ASV area under the concentration time curve (AUC) and subjects with baseline aspartate aminotransferase (AST) above 78 IU/L had a 58% increase in area under the concentration time curve (AUC). Asians subjects had a 46% higher steady-state AUC relative to White/Caucasian subjects. Other significant covariates were formulation, age, and gender.
Conclusion
The current PPK model provided a parsimonious description of ASV concentration data in HCV-infected subjects. Key covariates identified in the model help explain the observed variability in ASV exposures and may guide clinical use of the drug.
Funding
Bristol-Myers Squibb.
Similar content being viewed by others

Introduction
Chronic hepatitis C virus (HCV) infection, currently estimated to affect about 3% of world population, or 80–185 million people, frequently leads to hepatocellular carcinoma, cirrhosis, and liver transplantation [1, 2]. Asunaprevir (formerly BMS-650032, ASV) is a tripeptidic acylsulfonamide inhibitor of the HCV NS3/4A protease [3, 4]. Asunaprevir is approved for the treatment of chronic HCV in multiple countries as part of the all-oral direct-acting antiviral (DAA) DUAL combination regimen with the HCV NS5A inhibitor daclatasvir (DCV) or part of the QUAD regimen with DCV and peginterferon/ribavirin (pegINF/RBV) in patients with genotype 1 or genotype 4 HCV [5,6,7,8].
Asunaprevir has shown complex pharmacokinetic (PK) characteristics [9]. The dose–exposure relationship for ASV is generally non-linear. In multiple ascending dose (MAD) studies, ASV administered using an early capsule formulation resulted in greater-than-dose-proportional increases in exposure from 10 to 600 mg twice daily (BID) [10].
Asunaprevir is eliminated primarily via cytochrome P450 (CYP) 3A4-mediated hepatic metabolism, whereas distribution of asunaprevir to the liver occurs via organic anion-transporting polypeptide (OATP) [11,12,13]. Asunaprevir displays time-dependent nonlinear pharmacokinetics. Following multiple doses at 400 and 600 mg BID, ASV trough concentrations were lower on Day 14 compared to Day 1, suggesting auto-induction of CYP3A4. The magnitude of auto-induction appeared to be dose-dependent. At the 200 mg BID, the trough concentration ratio of Day 14 to Day 1 was 1.9, suggesting a more modest auto-induction. This is consistent with the weak inductive effect of asunaprevir on midazolam, a sensitive CYP3A4 probe substrate [13].
Hepatic function impairment had been shown to significantly impact the steady-state PK of ASV [14]. Asunaprevir exposures were approximately 5- to 10- and 20- to 30-fold higher in subjects with moderate (Child–Pugh B) and severe (Child–Pugh C) hepatic impairment, respectively, and as such ASV is not recommended to be used in these subjects. Asunaprevir exposure was also higher with increased aspartate aminotransferase (AST), alanine aminotransferase (ALT) and total bilirubin levels, and decreased albumin levels [14]. Furthermore, cross-study comparisons of steady-state ASV exposures from subjects with HCV infection indicated approximately 2- to 3-fold higher exposure than those from healthy subjects [9]. Inflammation, tissue damage and fibrosis associated with chronic HCV infection could affect liver uptake of ASV and/or reduce hepatic CYP3A4 activity, resulting in decreased clearance of the drug [15].
Ethnic differences have been reported in ASV exposure. In Phase II studies, ASV maximum concentration (Cmax) and AUC in Japanese subjects were approximately double compared to White subjects [16]. Data in Chinese and Indian subjects also suggested that exposure in these populations was closer to that observed in Japanese subjects than observed in Caucasian subjects. Body weight may be a contributing factor; however, there was a high degree of overlap in body weights between the Asian and White subjects in these clinical studies.
Significant food effects has been observed with a tablet formulation used in Phase I/II studies [9]. A soft-gel capsule, developed as a food-effect-mitigating formulation, is able to provide significantly higher exposures (approximately 4- to 5-fold for Cmax and 2-fold for AUC) with or without food than the tablet formulation given with food [17]. Subsequently, the soft-gel capsule was used in Phase III studies without food restrictions and is the commercial formulation [5].
A population PK analysis was conducted using data pooled from select Phase II/III studies to help better understand the complex PK profile and the exposure of ASV in subjects with chronic HCV infection at clinically relevant doses. More importantly, the analysis helps explain the sources of variability in ASV exposures by investigating potential relationships between covariates and PK parameters of ASV in the target patient population. The model would also provide individual post hoc predicted exposure values for subsequent exposure–response analyses.
Methods
Clinical Studies and Patient Population
Asunaprevir concentration, time data were obtained from 3 Phase II and 2 Phase III studies, among which 1 Phase II and 1 Phase III studies were in Japanese subjects (Supplemental Table 1 in the Electronic Supplementary Material). Asunaprevir was administered either as part of the DUAL regimen with DCV or as part of the QUAD regimen with DCV and pegINF/RBV. The asunaprevir doses investigated were 100, 200 and 600 mg, given once daily (QD) or BID, either in tablet or soft-gel formulation. Eligible subjects were male or female ≥ 18 years of age, with chronic HCV genotype 1 and 4 infections. Subjects with compensated cirrhosis and mild hepatic impairment were included. Subjects could be treatment naive, partial, null responders or ineligible/intolerant to the pegINF/RBV-based therapy.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Population Pharmacokinetic Analysis
Model Development
The population PK model development was comprised of establishing a base, full and final model. The base structural model development was guided by the graphical evaluation of ASV plasma concentration–time profiles. Also investigated as part of the base model were fixed-effect parameters associated with ASV characteristics known to influence its PK: the effect of auto-induction on ASV clearance (CL/F) and the effect of formulation. Inter-individual variability for the PK parameters was modeled assuming a log-normal distribution. Residual variability was investigated using either the proportional or the combined proportional plus additive error model for non-transformed concentrations, and additive error model for log-transformed concentrations. Model selection was based on significant reduction in the objective function value (OFV; p < 0.05), precision and plausibility of parameter estimates, and goodness-of-fit plots.
Once the structural base model was identified, covariate evaluations were performed to assess the influence of key patient and treatment characteristics on the PK of ASV. Covariate model building was conducted with the full model approach [18]. Covariates were added to the base model in a univariate fashion; and significant covariate-PK relationships were evaluated using the log-likelihood ratio test (LRT). All significant (p < 0.05) covariates were included in the full model. The selection of covariates and likely impacted PK parameters was based on clinical interest, pharmacological plausibility, prior knowledge and the availability of data. As baseline AST and ALT were highly correlated (r2 = 0.86), only the most significant term was kept in the full model if both covariates were found to be significant in the univariate screening. On-treatment AST and ALT values were also investigated as time-varying covariates on ASV CL/F, as treatments would normalize hepatic functions. Asunaprevir dose was investigated as a covariate on the relative bioavailability (F) based on the observed nonlinear dose–exposure relationship in the MAD study.
The final model was obtained using a stepwise backward elimination process to reach a parsimonious model. The covariate with the smallest change in objective function was removed from the model, and the process repeated until all remaining covariates were significant at p < 0.001 based on LRT. Uncertainties in the final model parameter estimates were reported with stratified nonparametric bootstrap 95% confidence intervals (CI) [19]. The final model was used for model application and for providing individual post hoc predicted exposure values for exposure–response analyses.
Model Evaluation
Model evaluation was performed using goodness-of-fit diagnostic plots, prediction corrected visual predictive checks (pcVPC) and shrinkage. As a graphic diagnostic tool, pcVPC was used to identify model misspecification, especially with respect to random effects, by comparing the observed ASV concentration–time profiles with the simulated median and distribution (5th to 95th percentile) [20]. A total of 1000 trial replicates were simulated using actual covariates and dose regimens, the final model parameter estimates, and simulated subject-specific random effects and residual errors. Shrinkage was assessed for inter-individual variability (η) and for residual error (ε). High η shrinkage (e.g., > 30%) would indicate lack of information for a reliable estimate of the inter-individual variability [21].
Model Application
The final model was used to investigate the effects of covariates on the PK of ASV. The impact of significant covariates on ASV population PK parameters was examined using forest plots. Pharmacokinetic parameters, determined at the 5th and 95th percentiles of a continuous covariate, or at different levels of a categorical covariate, were compared with typical PK estimates. Effect of covariate at the extreme values and associated 95% CI, when wholly contained within the 80–125% boundaries of the typical PK estimates, would suggest a lack of clinical relevance. The impact of significant covariates on ASV PK exposures at steady-state was also evaluated using simulation. All subjects were simulated to receive the 100-mg BID soft-gel Phase 3/commercial formulation, except when evaluating the effect of formulation, where subjects were simulated to receive their actual formulation at 100 mg BID. The contribution of each covariate independently to the overall variability of ASV PK exposures at steady-state was determined using the 5th and 95th percentile values of a continuous covariate, or at different levels of a categorical covariate, while fixing other covariates to their respective typical values in the population. The results were illustrated using tornado plots. Furthermore, ASV steady-state exposures were determined using all observed covariate values in the dataset and were summarized by quantiles of continuous covariates or levels of categorical covariates to allow the assessment of the impact of each covariate while taking into account potential correlations among covariates in the study population.
Analysis Platforms
The population PK analysis was performed using NONMEM (v.7.1), compiled using Intel FORTRAN Compiler (v.10.1; Intel). The first-order conditional estimation method was used throughout the model-building process. Perl-speaks-NONMEM (PsN, v.3.2 or later, http://psn.sourceforge.net/) was used to aid the model development. Diagnostic graphics, exploratory analyses, and post-processing of NONMEM output were performed using S-Plus (v.8.1;, TIBCO Software, Palo Alta, CA, USA), or R (v.3.0 or later, http://www.r-project.org).
Results
A total of 9496 ASV concentration–time data from 1236 subjects were included in the population PK analysis dataset. The majority of subjects had 4–9 samples. Key demographics and baseline characteristics are summarized in Table 1. The majority of subjects were White (58.3%) or Asian (34.2%, among which 21.5% were Japanese). The average baseline values for AST and ALT were 62 and 73 U/L, respectively, which were approximately two times the upper end of the normal range. Average ALT and AST values decreased by approximately 50% and were approaching the upper end of the normal range over the initial 6 weeks. Most subjects (69.7%) received the soft-gel capsule formulation at 100 mg BID as part of the DUAL regimen.
Structural Model
The final structural model was a two-compartment model with a zero-order release from the formulation followed by first-order absorption into the central compartment and first-order elimination. The auto-induction of ASV CL/F was modeled with a step function occurring 2 days after the initial dose. A change point at approximately 6 days after the initial dose was also investigated, but found to result in a greater OFV. Inter-individual random effects were included as a diagonal block on CL/F, central (Vc/F) and peripheral (Vp/F) volume of distribution, and the absorption rate constant (Ka). Residual variability was modeled using an additive error model with log-transformed ASV concentrations.
Covariate Models
Following the univariate covariate search process, age, weight, gender, race, baseline AST, ALT and creatinine clearance, cirrhosis, patient type, and host CC genotype were found to be significant covariates on CL/F whereas age, gender, baseline AST and ALT, and cirrhosis were significant for Vc/F. Additionally, weight, formulation and ASV dose were significant covariates for Vp/F, Ka and F, respectively. As the inclusion of baseline AST resulted in greater reduction in the OFV, baseline ALT were excluded from subsequent evaluations. All other variables were included in the full model. Following the stepwise backward elimination process, covariates excluded from the final model were weight on CL/F, creatinine clearance on CL/F, patient type on CL/F, host CC genotype on CL/F, age on Vc/F, and baseline AST and AST ratio on Vc/F. The final model did not converge due to rounding errors; however, bootstrap means were similar, and 95% confidence intervals included each parameter estimate. The final model parameters are provided in Table 2. The covariate–PK parameter relationships in the final model are shown as follows:
Model Evaluations
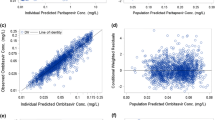
Goodness-of-fit diagnostic plots show good agreement between the predicted and the observed ASV concentrations, and that conditional-weighted residuals are unbiased over time and population predicted concentrations (Fig. 1). The random effect for CL/F, Vc/F, Vp/F and Ka showed a symmetric, bell-shaped distribution that was centered near zero. The pcVPC plots in Fig. 2 showed good agreement between the median of the data and the predicted medians and distributions; the variability appears to be slightly under-predicted in the first 4 h, and slightly over-predicted around 12 h. However, Fig. 2b, c shows consistency of the model with the pre- and post-induction periods with the 200-mg tablet. The shrinkage value for the residual error was 11%, indicative of the model’s adequacy in providing individual predictions for subsequent exposure–response analysis. Shrinkage on CL/F was 13%, suggesting each subject’s CL/F is well estimated and can be used for further exploration of covariate-CL/F relationships. Shrinkage values on Vc/F, Ka and Vp/F were 39, 42 and 65%, respectively, suggesting significant amounts of shrinkage in the empirical Bayes estimates of posterior individual parameters.

Goodness-of-fit diagnostic plots for the final model. CWRES conditional weighted residual. Dashed lines the lowest (local regression smoother) trend lines

Prediction-corrected visual predictive check plots for the final model. Circles are observed ASV plasma concentrations, and dashed red lines represent 5th, 50th and 95th percentiles of the observed values. The blue field represents 95% CI of the model simulated median and the red fields 95% CI of the 5% tile (lower) and 95% tile (upper) of the simulation-based prediction intervals. Data and predicted values are binned, and plotted at the center points of each bin. a All studies (n = 1236); b subjects who received the tablet formulation at 200 mg BID, pre-induction (n = 94); c subjects who received the tablet formulation at 200 mg BID, post-induction (n = 286). A corresponding pcVPC plot for the soft-gel formulation was not produced due to the lack of PK samples during the first week of dosing when auto-induction presumably developed
Effect of Covariates on Population PK Parameters
The impact of covariates retained in the final model on the population PK parameters is shown in Fig. 3. Asunaprevir CL/F decreased with increasing baseline AST, increasing AST relative to baseline, and cirrhosis, with the 95% CIs for these effects lay outside the 80–125% region. The CL/F decreased with increasing age and decreased in Asians compared to Caucasians, with the 95% CIs overlapped the 80–125% region. The effect of gender on CL/F lay within the 80–125% boundaries. Relative bioavailability was lower in subjects receiving the tablet relative to the soft-gel formulation, but increased with the 600-mg tablet dose relative to the other tablet doses. The duration of absorption increased with the tablet formulation. The effect of covariates on other population PK parameters should be interpreted with caution given the high shrinkage values associated with these parameters.

Effect of covariates on asunaprevir population pharmacokinetic parameters of the final model. PK parameters were referenced to a non-cirrhotic, male, 70-kg, 55-year-old White subject with baseline AST of 60 IU/L receiving the soft-gel prior to induction. The horizontal axis represents percent change in PK parameter relative to the reference. The thick horizontal lines indicate the PK parameter change at the 5th and 95th percentile of the continuous covariates. The points indicate the PK parameter change attributed to the respective continuous covariate value or categorical covariate. The thin horizontal lines represent the 5th to 95th percentile confidence interval in the estimated value. Vertical lines indicate the reference PK parameter value (100%) and 80% and 125% changes
Effect of Covariates on ASV Steady-State Exposures
The impact of each covariates independently on ASV steady-state AUC based on the final model is shown in Fig. 4a. Baseline AST, cirrhosis, and change in AST from baseline had moderate effects, each covariate contributing 30–50% change in AUC. The effect of Asian race was modest; Asians have a 29.2% increase in AUC relative to the White/Caucasians. Other covariates such as age, gender, and weight had a minimal effect (< 20%). The predicted steady-state AUC was 19% lower with the tablet than with the soft-gel formulation, which was less than expected given known differences in relative bioavailability between the two formulations. A similar trend was observed for ASV steady-state minimum concentration (Cmin), although the magnitude of covariate effects was generally larger (Fig. 4b).

Effect of covariates on asunaprevir steady-state exposures. The black bar represents the 5th to 95th percentile range of the exposures calculated using individual PK parameter estimates. The impact of each covariate on exposure was calculated using the 5th to 95th percentile range of each continuous covariate, or the covariate category, while fixing other covariates to the respective typical values in the population
The impact of select covariate on ASV steady-state exposures when taking into account the potential correlations amongst covariates in the patient population is shown in Fig. 5 and Table 3. Asunaprevir steady-state AUC increased from − 25.6 to 57.7% relative to the median value in subjects with baseline AST from the first to the fourth quartile (Fig. 5a). Cirrhotic subjects had an 84.3% increase in AUC relative to non-cirrhotic subjects (Fig. 5b). Japanese and non-Japanese Asians had an approximately 40–45% increase in AUC relative to the White/Caucasian subjects (Fig. 5c). The tablet formulation (100 mg BID with actual observed patient characteristics in the dataset) produced an approximately 40% lower AUC compared to the soft-gel formulation (Fig. 5d). The ASV steady-state AUC changed more modestly with age, gender and weight, suggesting a lack of clinical significance (data not shown).

Effect of select covariates on asunaprevir steady-state AUC. Steady-state exposure was calculated using individual PK parameter estimates. The box shows the median, 25th and 75th percentile per group. The whiskers extend to 1.5 times the interquartile range. The notch indicates an approximate 95% confidence interval on the median, and is calculated as 1.58 times the interquartile range normalized to the number of data points
Discussion
The purpose of this analysis was to characterize the population PK of ASV in patients with chronic HCV infection, who are treatment-naive, non-responders and ineligible/naive intolerant to IFN-based therapy. This analysis provides a quantitative description of ASV PK and, in particular, evaluated the impact of key covariates such as hepatic function, race, gender, age, weight and formulation on ASV steady-state exposures. A two-compartment model with first-order elimination from the central compartment, an induction effect on clearance, and an absorption model consisted of zero-order release followed by first-order absorption adequately described ASV PK after oral administration. For a typical White, non-cirrhotic 70-kg, 55-year-old male with baseline AST of 60 IU/mL receiving the 100 mg BID of the soft-gel formulation, ASV CL/F was 50.8 L/h, increasing by 43% after 2 days to a CL/F of 72.5 L/h at steady-state, most likely due to auto-induction of CYP3A4. Since there were limited ASV PK data available during the first 7 days after the start of dosing in the PPK dataset, the dynamics of the induction process could not be estimated. Therefore, a change in clearance with time was investigated with a step function as part of the base model.
Factors indicative of hepatic function were identified as key influential covariates on ASV exposures. Subjects with cirrhosis had an approximately 84% increase in ASV AUC (Fig. 5b) and subjects with baseline AST in the fourth quartile (above 78 IU/L) had an approximately 58% increase in AUC (Fig. 5a). Baseline ALT was also identified as a significant covariate on ASV CL/F in the univariate screening step. However, it was not included in the final model due to its high correlation with baseline AST. These results are consistent with the pharmacokinetic profile of ASV which is transported into the liver by OATP1B1, and is extensively metabolized in the liver via the CYP3A4-mediated oxidative pathway and eliminated primarily through the feces. The effect of AST on CL/F has been previously reported for drugs that are extensively metabolized in the liver, such as sildenafil and tacrolimus [22, 23]. Cirrhosis changes the architecture of the liver leading to changes in blood flow, protein binding and drug-metabolizing enzymes [24]. Reduction in the enzyme level and activity due to the loss of hepatic function could result in decreased clearance of drugs that are primarily metabolized by the liver. In the hepatic impairment study conducted in non HCV-infected subjects, ASV exposures were significantly higher in subjects with moderate and severe hepatic impairment. In the hepatic impairment study, there were no significant changes in ASV exposure in subjects with mild hepatic impairment compared to matched controls with normal hepatic function, whereas ASV AUC was 10- and 32-fold higher in subjects with moderate and severe hepatic impairment, respectively. The results from the population PK analysis therefore provided further evidence that ASV exposures were highly influenced by the degree of hepatic impairment. Worsening of hepatic function may lead to increased ASV exposures, which may in turn impact the safety profile of ASV. However, it should be noted that, in the current dataset, ASV exposures in subjects with cirrhosis and those with elevated AST levels were limited to < 2-fold of the population median exposure value, and were well within the range of observed exposures in Phase II/III studies. This modest change is consistent with the fact that only subjects with compensated cirrhosis and hepatic function no worse than Child–Pugh A (mild impairment) were included in the Phase II/III population. Therefore, a potential limitation of the model is to extrapolate ASV exposures in patients with cirrhosis and worsening hepatic function (Child–Pugh B and C). In addition, it is known that ASV exposures are generally higher in subjects with chronic HCV infection compared to those in healthy subjects, highlighting the role of hepatic function in the PK of ASV. Although the number of subjects with cirrhosis was limited, the difference in plasma exposure in subjects with compensated cirrhosis did not appear to be clinically meaningful, as virological outcomes in these subjects were comparable to those in subjects without cirrhosis. In Phase III studies with the DUAL regimen, only 3% of subjects had experienced Grade 3/4 AST or ALT elevations, consistent with findings from a previous exposure–response analysis that suggested minimal increase in the probability of liver safety events, with an approximate doubling of ASV AUC at the clinically relevant dose [25, 26]. On the other hand, subjects receiving the DUAL treatment had in general reduced AST levels as a result of viral clearance. Covariate effect of the time-varying AST change from baseline suggested that, for subjects with the largest reduction in AST during treatment (43% decrease from baseline), ASV CL/F increased by 27%. As the subjects’ hepatic function normalized, there was a lowering effect on ASV exposure which thus further mitigated the risk of safety events.
Race was identified as a significant covariate on ASV CL/F, with approximately 29% lower CL/F in Asian subjects when race was assessed as an independent covariate. However, when the effect of race on ASV AUC was evaluated after accounting for potential correlations between covariates, Asians in general and Japanese subjects had approximately 46 and 55% higher steady-state AUC and Cmin, respectively, relative to the White/Caucasian subjects. The overall higher ASV exposures in Asians and Japanese subjects were likely contributed by the different age and gender distribution among the study populations. In the current dataset, approximately 21.5% subjects were Japanese, and Japanese subjects had a higher proportion of females (approximately 65% compared to 37%) and higher median age (approximately 62 years compared to 55 years) than non-Japanese subjects. Advanced age had been estimated to independently contribute to approximately 9 and 16% higher ASV steady-state AUC and Cmin, respectively (Fig. 4). Females were estimated to have 12 and 19% higher ASV steady-state AUC and Cmin, respectively.
The effect of formulation on the bioavailability of ASV suggested that the bioavailability of the Phase III/commercial soft-gel capsule formulation was approximately 1.3-fold higher than the tablet formulation used in the Phase II studies. Based on the relative bioavailability study in healthy subjects, it was expected that ASV bioavailability following the soft-gel Phase III/commercial formulation at 100 mg BID would be approximately 2-fold higher relative to the Phase II tablet at 200 mg BID. In the current dataset, subjects received a wide range of the tablet doses/regimens including 200 mg QD, 200 mg BID, 600 mg QD and 600 mg BID. As reported previously [9], and identified in the current analysis, ASV exposure increased more than proportionally with an increase in dose from 200 mg BID to 600 mg BID of the Phase II tablet, especially in Japanese subjects, which may have also contributed to the lower estimate of the difference in bioavailability between the soft-gel and tablet formulations. When ASV 100 mg BID of the soft-gel capsule was directly compared to the tablet at the same dose, simulations using the final model suggest that the tablet had approximately 40% lower AUC, which would translate into an approximate difference of 1.7-fold in bioavailability between the formulations, which was closer to the expected difference.
Other covariates such as age, gender, and weight which were retained in the final model had statistically significant effects; however, with marginal importance, i.e. < 20% effect on ASV steady-state exposure. In addition to the above-described covariates the full model identified the following covariates as significant (p < 0.05): sex, weight, patient type and CC genotype on CL/F and baseline AST and time-varying AST on Vc/F. These covariates were dropped from the final model during the backward elimination process. Other covariates, including OATP transporters and comedications, were not identified as significant in the univariate screening, and were not included in the full model, and therefore it can be inferred that they did not help explain the observed PK variability in ASV.
Conclusion
In conclusion, the current ASV PPK model provided a parsimonious description of the concentration data in subjects with chronic HCV infection. The key covariates identified and described in the model, such as hepatic function status, race and formulation, had statistical and biological plausibility that help explain the observed variability in ASV exposures in the included Phase II/III studies and may guide clinical use of the drug. The results of the analysis were used for the development of the exposure–response analyses.
References
Gower E, Estes CC, Hindman S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61(S1):S45–57.
Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556–62.
McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, et al. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother. 2012;56:5387–96.
Gentile I, Buonomo AR, Zappulo E, Minei G, Morisco F, Borrelli F, Coppola N, Borgia G. Asunaprevir, a protease inhibitor for the treatment of hepatitis C infection. Ther Clin Risk Manag. 2014;26(10):493–504.
Manns M, Pol S, Jacobson IM, Marcellin P, Gordon SC, Peng CY, et al. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: a multinational, phase 3, multicohort study. Lancet. 2014;384:414–29.
Bronowicki JP, Ratziu V, Gadano A, Thuluvath PJ, Bessone F, Martorell CT, et al. Randomized trial of asunaprevir plus peginterferon alfa and ribavirin for previously untreated genotype 1 or 4 chronic hepatitis C. J Hepatol. 2014;61:1220–7.
Zeuli JD, Adie SK, Rizza SA, Temesgen Z. Drugs today (Barc), Asunaprevir plus daclatasvir for the treatment of chronic hepatitis C virus infection. Drugs Today (Barc). 2015;51(11):629–43.
Yang SS, Kao JH. Asunaprevir-containing regimens for the treatment of hepatitis C virus infection. Expert Rev Gastroenterol Hepatol. 2015;9(1):9–20.
Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug–drug interactions. Clin Pharmacokinet. 2015;54(12):1205–22.
Eley T, He B, Huang SP, Li W, Pasquinelli C, Rodrigues AD, Grasela DM, Bertz RJ. Pharmacokinetics of the NS3 protease inhibitor, Asunaprevir (ASV, BMS-650032), in phase I studies in subjects with or without chronic hepatitis C. Clin Pharmacol Drug Dev. 2013;2(4):316–27.
Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug–drug interactions. Clin Pharmacokinet. 2015;54:1205–22.
Gong J, Eley T, He B, Arora V, Philip T, Jiang H, Easter J, Humphreys WG, Iyer RA, Li W. Characterization of ADME properties of [(14)C]asunaprevir (BMS-650032) in humans. Xenobiotica. 2016;46(1):52–64.
Eley T, Gardiner D, Persson A, He B, You X, Shah V, et al. Evaluation of drug interaction potential of the HCV protease inhibitor asunaprevir (ASV; BMS-650032) at 200 mg twice daily in metabolic cocktail and P-glycoprotein (P-gp) probe studies in healthy volunteers [abstract]. Hepatology. 2011;54:abstract no. 381.
Eley T, He B, Chang I, Colston E, Child M, Bedford W, et al. The effect of hepatic impairment on the pharmacokinetics of asunaprevir, an HCV NS3 protease inhibitor. Antivir Ther. 2015;20(1):29–37.
Morcos PN, Moreira SA, Brennan BJ, Blotner S, Shulman NS, Smith PF. Influence of chronic hepatitis C infection on Asunaprevir Preclinical and Clinical PK and Drug Interactions 1221 cytochrome P450 3A4 activity using midazolam as an in vivo probe substrate. Eur J Clin Pharmacol. 2013;69:1777–84.
Eley T, Chan P, Huang S, Wind-Rotolo M, Delmonte T, He B, et al. Asunaprevir in Japanese subjects in phase 2: exposure-safety versus US/EU-based subjects and preliminary assessment of correlation with single nucleotide polymorphisms (SNPs) in liver uptake transporters. Hepatology. 2012;56(Suppl 1):1070A–1A.
Eley T, Chan P, Sverdlov O, He B, Bedford B, Kandoussi H, et al. Improved bioavailability and mitigated food effect for asunaprevir utilizing a lipid-based formulation: similar exposure with 100 mg twice-daily soft-gel capsule relative to 200 mg twice daily of phase 2 tablet [abstract/poster no. A-1247]. In: 52nd Interscience conference antimicrobial agents chemotherapy. San Francisco; 9–12 Sep 2012.
Marc Gastonguay. full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. Page 20 (2011) Abstr 2229. http://www.page-meeting.org/?abstract=2229. Accessed 21 Mar 2018.
Parke J, Holford NH, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Programs Biomed. 1999;59(1):19–29.
Bergstrand M, Hooker AC, Wallin JE, et al. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143.
Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558–69.
Milligan PA, Marshall SF, Karlsson MO. A population pharmacokinetic analysis of sildenafil citrate in patients with erectile dysfunction. Br J Clin Pharmacol. 2002;53(Suppl 1):45S–52S.
Oteo I, Lukas JC, Leal N. Tacrolimus pharmacokinetics in the early post-liver transplantation period and clinical applicability via bayesian prediction. Eur J Clin Pharmacol. 2013;69(1):65–74.
Elbekai RH, Korashy HM, El-Kadi AOS. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr Drug Metab. 2004;5(2):157–67.
Chan P, et al. Exposure-response analyses of asunaprevir in combination with daclatasvir ± peginterferon/ribavirin among patients with genotype 1 chronic HCV infection: dose selection for phase 3 clinical trials. J Hepatol. 2013;58(Suppl1):S328–9.
Gentile I, Zappulo E, Buonomo AR, Scotto R, Borgia G. Asunaprevir for hepatitis C: a safety evaluation. Expert Opin Drug Saf. 2015;14(10):1631–46.
Acknowledgements
Funding
The studies described in this report and the article processing charges were funded by Bristol-Myers Squibb.
Authorship
All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Yash Gandhi is an employee of Bristol-Myers Squibb Research and Development. Tushar Garimella is an employee of Bristol-Myers Squibb Research and Development. Li Zhu is an employee of Bristol-Myers Squibb Research and Development. Richard Bertz is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Richard Bertz is currently employed at University of Pittsburgh (Pittsburgh, PA, USA). Marc Bifano is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Marc Bifano is now retired. Phyllis Chan is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Phyllis Chan is currently employed at Genentech Inc (South San Francisco, CA, USA). Timothy Eley is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Timothy Eley is currently employed at Arbutus Biopharma Corporation (Warminster, PA, USA). Eric Hughes is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Eric Hughes is currently employed at Novartis International AG (Basel, Switzerland). Malaz AbuTarif is a former employee of Bristol-Myers Squibb and was employed by Bristol-Myers Squibb when the studies and analyses were conducted. Malaz AbuTarif is currently employed at Daiichi Sankyo Inc (Parsippany, NJ, USA). Mayu Osawa is an employee of Bristol-Myers Squibb K.K. Takayo Ueno is an employee of Bristol-Myers Squibb K.K. Hanbin Li is currently employed by Certara and was funded by Bristol-Myers Squib to carry out the modeling analyses reported in this paper.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to https://doi.org/10.6084/m9.figshare.5966257.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhu, L., Li, H., Chan, P. et al. Population Pharmacokinetic Analysis of Asunaprevir in Subjects with Hepatitis C Virus Infection. Infect Dis Ther 7, 261–275 (2018). https://doi.org/10.1007/s40121-018-0197-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-018-0197-y