
Abstract
Cardiovascular diseases are the leading cause of death worldwide. Research in the last two decades has emphasized the inflammatory process as a key component in the pathogenesis of many of them. The Interleukin-1 family is a pivotal element of inflammation and has been well studied as a therapeutic target in various inflammatory states. Recent trials have explored the effect of Interleukin-1 blockade in cardiovascular diseases and initial evidence of the relevance of such treatment in this field of medicine accumulate. This review will describe the role of Interleukin-1 in heart diseases and the potential therapeutic effect of its blockade in such diseases.
Similar content being viewed by others

Introduction
The inflammatory process, which is the biological response to tissue injury, was found to have a major role in the mechanism of different cardiac pathologies. The inflammatory cascade is initiated at the site of local injury and is transmitted by a variety of mediators known as cytokines. The Interleukin-1 (IL-1) family of ligands and receptors is the main cytokine family associated with acute and chronic inflammation. Among the various members of the IL-1 family, IL-1β has been proven to be a therapeutic target for a growing number of autoinflammatory diseases. Suppression of IL-1β results in a prompt and persistent decrease in disease severity [1].
Inflammation plays a central role in many cardiovascular diseases, including heart failure [2], myocardial infarction [3], arrhythmias [4], pericarditis [5], myocarditis [6], and sepsis-induced cardiomyopathy [7]. Interest in implementation of anti-inflammatory treatment in the cardiovascular field, targeting the IL-1 family of ligands and receptors, has led to different trials in recent years.
Thorough and comprehensive reviews dealing with targeting IL-1 in heart diseases have been published in recent years [8, 9]. This narrative review will discuss the role of IL-1 in the pathogenesis of different heart diseases, the potential use of IL-1 blockade to improve clinical outcomes in these diseases, and will include updated results from different recent clinical trials.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
The IL-1 Family
Understanding the role of IL-1 in the pathogenesis of disease has improved greatly since the publication of “Biologic Basis for Interleukin-1 in Disease” [10]. Blocking IL-1β is now the standard of care for a class of autoinflammatory diseases [11]. Autoinflammatory conditions are often responsive to IL-1β blockade, and much less responsive to immunosuppressive therapies, which are more suitable for autoimmune diseases, in which the T cell is the “key player” of inflammation [1].
At first, only two forms of IL-1 were recognized, IL-1α and IL-1β. During the following years, the IL-1 family has expanded and now includes 11 ligands and another set of receptors, co-receptors and inhibitory receptors. Most of the ligands are pro-inflammatory while the others serve as antagonists and regulate the inflammatory response. One ligand (IL-37) is even anti-inflammatory [1].
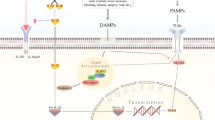
The biological activity of IL-1 remains the model for mediating inflammation (Fig. 1). IL-1α or IL-1β bind first to their receptor, termed type 1 (IL-1R1). Then the co-receptor chain, termed the accessory protein (IL-1RAcP), is recruited. This triple complex recruits the adaptor protein MyD88 to the Toll-IL-1 receptor (TIR) domain of each receptor. Several kinases are phosphorylated, nuclear factor-κB (NF-κB) translocates to the nucleus, and the expression of a vast collection of inflammatory genes, including IL-1, ensues [12]. Another key player is the “inflammasome”, a cytosolic molecular structure composed of an adaptor protein, pro-caspase 1, and a sensor molecule. The best-characterized inflammasome has a sensor molecule called nucleotide-binding domain and leucine-rich repeat pyrin domain containing 3 (NLRP3). This sensor molecule may be activated by both infectious stimuli, known as pathogen-associated molecular patterns (PAMPs), and sterile stimuli, known as damage-associated molecular patterns (DAMPs), such as cholesterol, amyloid-beta, urate crystals, and more. This activation is based on either ATP binding to the P2X7 receptor and a secondary efflux of potassium to the extracellular space or reactive oxygen species (ROS) formation. Upon activation of the NLRP3-inflammasome, pro-caspase-1 is converted to an active enzyme. Active caspase-1 then cleaves the IL-1 precursor in specialized secretory lysosomes or in the cytosol, followed by secretion of “mature” IL-1β [1, 13].

IL-1β synthesis and secretion. IL-1β binds to the IL-1 receptor type 1 (IL-1R1). Then the co-receptor chain, termed the accessory protein (IL-1RAcP), is recruited. This triple complex recruits the adaptor protein MyD88 to the Toll-IL-1 receptor (TIR) domain. Several kinases are phosphorylated, nuclear factor-κB (NF-κB) translocates to the nucleus, and pro-ILβ transcription ensues. The NLRP3-inflammasome is a cytosolic molecular structure composed of an adaptor protein, pro-caspase 1, and the NLRP3 sensor molecule, which may be activated by both infectious stimuli, known as pathogen-associated molecular patterns (PAMPs) and sterile stimuli, known as damage-associated molecular patterns (DAMPs). This activation is based on either ATP binding to the P2X7 receptor with a secondary efflux of potassium to the extracellular space or reactive oxygen species (ROS) formation. Upon activation of the NLRP3-inflammasome, pro-caspase-1 is converted to an active enzyme. Active caspase-1 then cleaves the IL-1 precursor in specialized secretory lysosomes or in the cytosol, followed by secretion of “mature” IL-1β
Blocking IL-1-Mediated Disease
Anakinra is a recombinant human interleukin-1 competitive receptor antagonist that blocks the biologic effects of interleukin-1, thereby reducing systemic inflammatory responses. In 2001, anakinra received US Food and Drug Administration approval for the treatment of rheumatoid arthritis. It has also been successfully studied in randomized trials as an effective treatment option for other systemic autoinflammatory diseases, such as Familial Mediterranean fever (FMF), cryopyrin-associated periodic syndromes (CAPS), TNF receptor-associated periodic syndrome (TRAPS), adult and juvenile Still’s disease, and others. Today, some of these diseases are also successfully treated with neutralization by human anti–IL-1 monoclonal Abs, such as canakinumab, which is an approved treatment for FMF, CAPS, TRAPS, and more.
IL-1 in Heart Failure
Patients with chronic or decompensated heart failure (HF) demonstrate a marked increase in a variety of proinflammatory cytokines, including IL-1, with increasing levels according to the degree of disease severity, independent of whether the etiology is ischemic, hypertensive, idiopathic dilated cardiomyopathy, or inflammatory [14]. The inflammatory biomarker C-reactive protein (CRP), which is a known surrogate marker for IL-1 activity, is an independent predictor of adverse outcomes in patients with acute or chronic heart failure [2]. The cytokine hypothesis of heart failure suggests that a precipitating event triggers activation of proinflammatory cytokines, which leads to detrimental effects on left ventricular function and accelerates the progression of heart failure.
Some mechanisms relating IL-1 to impaired systolic function have been purposed. IL-1β was shown to decrease the beta-adrenergic responsiveness of L-type calcium channels in a cAMP-independent mechanism [15]. Moreover, IL-1β decreased the expression of genes important to the regulation of calcium homeostasis (phospholamban, sarcoplasmic reticulum calcium ATPase), in a reversible manner [16]. IL-1β also increases nitric oxide synthase (NOS) expression in cardiac myocytes, leading to increased nitric oxide (NO) activity, lower energy production, and myocardial contractility, through a direct attack on the mitochondria [17, 18].
Some members of the IL-1 family have favorable cardiac effects. Two family members, IL-33 and ST2, are a ligand and a receptor that combine to enter the cardiac myocyte and protect against hypertrophy and fibrosis. ST2 exists as two main isoforms, a transmembrane receptor ST2L and a soluble receptor sST2. During adverse conditions such as wall stress, fibrosis, and inflammation, sSt2 is shed and acts as a decoy receptor for IL-33, blocking the favorable effects of IL-33 in the heart [19]. Increased levels of sST2 are expressed in patients with cardiac hypertrophy, fibrosis, ventricular dilatation, and reduced ventricular contractility, and is considered an independent predictor of 1-year mortality in acute decompensated HF [20]. It was also proved to be a strong predictor of hospitalizations and mortality in chronic, well-treated, stable CHF patients [21]. Patients with elevated sST2 levels tend to have abnormal echocardiographic findings, including enlarged left ventricular volumes, reduced contractility, and increased pressures. Thus, several studies have determined that those with chronic heart failure seem to have better hemodynamic profiles when sST2 is below 35 ng/ml. As a result, it was suggested that outpatient therapies should be aimed at this new therapeutic threshold goal [22].
Animal models describe a reversible systolic dysfunction and reduced LV contractility reserve (measured by a reduced responsiveness to isoproterenol) following a single or multiple injections of IL-1β in otherwise healthy mice [23]. Exploring the effect of circulating IL-1 activity, mice were injected with plasma obtained from patients with acute decompensated systolic HF, patients with stable chronic systolic HF, as well as from healthy subjects. The results were similar to exogenous administration of IL-1β, as described above—plasma from decompensated HF patients (but not from healthy controls) induced a significant systolic and diastolic dysfunction and reduced contractile reserve. Interestingly, mice who were pretreated with anakinra or an IL-1β antibody avoided this deleterious effect [24], which led to the assumption that IL-1β acts as a cardio-depressant, as was shown in severe sepsis [25]. Mice injected with plasma from patients with stable chronic systolic HF with elevated plasma levels of CRP showed normal resting systolic function but significantly impaired contractile reserve [24].
In the cardio-oncology field, one study in mice showed that IL-1 mediates the doxorubicin cardiotoxicity and a sequential study approved that blocking IL-1 with anakinra in such mice diminished doxorubicin-induced microstructural damages of cardiac tissue and rescued doxorubicin-caused reduction of cardiac functions exemplified by left ventricle ejection fraction (LVEF) and fraction shortening [26]. In fact, IL-1 was also found to play a role in radiation-induced cardiomyopathy, with mice divided into wild-type vs. IL-1R1 knock-out, and exposed to thoracic X-ray therapy. Knock-out mice, and wild-type mice treated with anakinra, showed preserved contractile reserve and LVEF at 4 months, in oppose to wild-type mice without treatment, which showed impairment in both parameters. Interestingly, all groups did not avoid other deleterious effects, such as left ventricle (LV) end-diastolic pressure, associated with increased myocardial interstitial fibrosis and pericardial thickening [27].
The first clinical study to evaluate the effects of IL-1 blockade on cardiac function reported that a single injection of anakinra (150 mg) in patients with rheumatoid arthritis (RA) without HF significantly improved parameters of myocardial contractility and relaxation, coronary flow reserve (measured by echocardiography), and endothelial function (measured by brachial artery flow-mediated dilatation). These were observed within 3 h of treatment and were sustained after 1 month of treatment [28]. A case report of a patient with RA and heart failure with preserved ejection fraction (HFpEF) who improved her NYHA functional class and peak aerobic capacity after replacing Etanercept, a TNF inhibitor, with anakinra, also implied the beneficial effects of IL-1 blockade on HF [29].
The AIR-HF was a pilot clinical study that tested the effect of 2 weeks of treatment with anakinra on cardiopulmonary exercise performance in seven patients with heart failure with reduced ejection fraction (HFrEF) and evidence of elevated CRP (> 2 mg/L). Both median peak oxygen consumption (VO2) and ventilator efficiency (VE/VCO2 slope) improved from baseline, both being statistically significant [24].
The ADHF study randomized 30 patients with acute decompensated HF, reduced LVEF (40%), and elevated CRP levels (≥ 5 mg/l) to either receive anakinra 100 mg twice daily for 3 days followed by once daily for 11 days or matching placebo, in a 1:1 double blinded fashion. At 72 h, anakinra reduced CRP by 61% versus baseline, compared with a 6% reduction among patients receiving placebo (P = 0.004). In patients with paired baseline and day 14 echocardiograms (n = 14), anakinra was associated with a greater recovery in LVEF [+ 10% (+ 3, + 14)] compared with placebo [0 (− 16% to + 5%), P = 0.020]. There were no significant differences between treatment groups in the initial length of stay or total hospital days during the 14 days. Two patients in the anakinra group and three patients in the placebo group experienced worsening HF or readmission for HF [OR 0.61 (0.09–4.34), P = 0.62] [30].
In the D-HART trial, a total of 12 patients with HFpEF with NYHA class II–III symptoms were enrolled in a double-blind, randomized, placebo-controlled, crossover trial and assigned 1:1 to receive anakinra 100 mg or placebo for 14 days and an additional 14 days of the alternate treatment (placebo or anakinra). A cardiopulmonary exercise test was performed at baseline, after the first 14 days, and after the second 14 days of treatment. The primary end point was placebo-corrected interval change in peak oxygen consumption. All 12 patients completed both phases and experienced no major adverse events. Anakinra led to a statistically significant improvement in peak oxygen consumption (+ 1.2 ml/kg/min, P = 0.009) and a significant reduction in plasma CRP levels (− 74%, P = 0.006). The reduction in CRP levels correlated with the improvement in peak oxygen consumption (R = − 0.60, P = 0.002) [31].
IL-1 blockade in the Recently Decompensated Heart Failure Anakinra Response Trial (REDHART) will evaluate the safety and efficacy of anakinra in adult patients with recently decompensated heart failure and systolic dysfunction (LVEF < 50%), in terms of aerobic exercise capacity and ventilatory efficiency measured by a cardiopulmonary exercise test. Secondary outcomes of survival free of hospital admissions will be tested. This randomized, double-blinded, placebo-controlled trial enrolled 60 patients who were treated within 2 weeks after discharge with anakinra 100 mg daily for 2 weeks, anakinra 100 mg daily for 12 weeks, or placebo daily for 12 weeks with a follow-up of 24 weeks. Results have yet to be published.
The D-HART 2 trial is a phase 2, 2:1 randomized, double-blind, placebo-controlled, single-center clinical trial, which enrolled HFpEF patients, NYHA class II-III symptoms, with hsCRP levels > 2 mg/l, to receive anakinra 100 mg once daily or placebo for 12 weeks. The co-primary endpoints will be placebo-corrected interval changes in peak oxygen consumption and ventilatory efficiency at week 12, and the secondary endpoints will investigate the effects of IL-1 blockade on cardiac structure, systemic inflammation, endothelial function, quality of life, body composition, nutritional status, and clinical outcomes. The study was completed in June 2017 and results are awaited [32].
IL-1 in Coronary Artery Disease, Myocardial Infarction, and Remodeling
Inflammation plays a key role in coronary artery disease (CAD) and other manifestations of atherosclerosis [3]. IL-1 plays a crucial role in stimulation of the post-infarction inflammatory response and is involved in the pathogenesis of cardiac remodeling. Thus, targeting the IL-1 signaling cascade may be a promising therapeutic target for patients with myocardial infarction [33]. In a mouse model of myocardial infarction, IL-1α is released by dying cardiomyocytes [34] and IL-1β synthesis is significantly upregulated after infarction [35] and is predominantly localized in leukocytes, fibroblasts, and vascular cells, but also in cardiomyocytes [36].
For many years, the IL-1 family has been associated with the regulation of endothelial and smooth muscle cell mitogenesis, thrombogenic response of endothelial cells, leukocyte adherence, lipoprotein metabolism, extracellular matrix production, and vascular permeability [37]. It is also related to plaque formation and rupture via different pathways: stimulation of vascular smooth muscle through upregulation of growth factor-β (TGF-β) [38], suppression of endothelial cell proliferation [39], expression of adhesion molecules by endothelial cells [40], and by modification of endothelium, which later favors thrombosis [41]. Neutrophil elastase (NE) was found to be a potential “trigger” for IL-1 processing and release from human coronary endothelial cells [42].
Interleukin-37 (IL-37) is a member of the IL-1 family, which was found to correlate with severe coronary artery calcification (CAC) [43]. Patients with CAD have higher levels of IL-1RI compared with normal people. Within the CAD patients, those with myocardial infarction have the highest levels, compared with unstable angina (UA) or stable angina pectoris (AP) patients [44].
Genetic loss of IL-1R1 in mice decreases dilation of the infarcted heart, reducing collagen deposition and attenuating matrix metalloproteinases expression [45]. Anakinra has reduced cardiomyocytes apoptosis and inhibited caspase-1 and caspase-9 activities in a mouse and rat model of myocardial infarction [46]. Treating post-MI mice with specific anti-IL-1β antibodies had similar consequences, with prevention of deterioration in systolic and diastolic function [47].
A trial investigating the effect of anakinra on RA patients reported a greater improvement in coronary flow reserve, arterial compliance, and ejection fraction, in addition to apoptotic and oxidative markers in CAD patients compared to patients without CAD [48].
Small clinical trials have tested the effectiveness of IL-1 inhibition in patients with myocardial infarction. The VCU-ART trial randomized ten patients with ST-segment elevation MI (STEMI) to daily anakinra or placebo for 14 days, in a double-blind fashion. Anakinra was shown to be safe and presented an improvement in left ventricle volume indexes, both in cardiac magnetic resonance (CMR) and in echocardiography, on a 3-month follow-up [49]. The following VCU-ART 2 could not reproduce this effect in a larger population of 30 patients with stable STEMI. Nevertheless, combining the two cohorts yielded a significant reduction in incidence of new HF with anakinra (30 vs. 5%, P = 0.035) [50]. An extended follow-up, to a median of 28 months, has shown a significant reduction in new-onset HF without a significant effect on recurrent ischemic events [51]. The ongoing VCU-ART 3 has another arm of high-dose anakinra, with a primary endpoint of CRP levels in 14 days and secondary endpoints of LVEF and new-onset HF, all in 12 months of follow-up. Study completion is estimated to be in February of 2018.
The MRC-ILA Heart Study was a phase 2, double-blinded, randomized, placebo-controlled study that recruited 182 patients with NSTEMI presenting < 48 h from onset of chest pain, dividing them to receive daily anakinra or placebo for 14 days. Anakinra showed a reduction in inflammatory markers without infarct size effect on CMR. Major adverse cardiovascular event (MACE) at day 30 and 3 months was similar. The study showed a significant excess of events in 12 months of follow-up in the anakinra group, but was under-powered for late events [52].
The CANTOS study is a randomized, double-blind, placebo-controlled, multi-center trial of canakinumab treatment, involving 10,061 patients with previous MI and CRP levels of 2 mg or more per liter. The trial compared three doses of canakinumab (50, 150, and 300 mg, administered subcutaneously every 3 months) with placebo. The primary end point was nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death. At a median follow-up of 3.7 years, the incidence rate for the primary end point was 4.50 events per 100 person-years in the placebo group, 4.11 in the 50-mg group [HR 0.93 (95% CI 0.80–1.07; P = 0.30)], 3.86 in the 150-mg group [HR 0.85 (95% CI 0.74–0.98; P = 0.021)], and 3.90 in the 300-mg group [HR 0.86 (95% CI 0.75–0.99; P = 0.031]. The 150-mg dose, but not the other doses, met the prespecified multiplicity-adjusted threshold for statistical significance for the primary end point and the secondary end point that additionally included hospitalization for unstable angina that led to urgent revascularization—5.13 events per 100 person-years in the placebo group vs. 4.29 in the 150-mg group [HR 0.83 (95% CI 0.73–0.95; P = 0.005). A 15% relative risk reduction of the primary end point with the 150-mg dose of canakinumab translates to an absolute risk reduction of 0.64% or a number needed to treat (NNT) of 156. Nearly all of that reduction came in nonfatal MI. There was no significant difference in stroke, cardiovascular death, and overall mortality. A secondary analysis showed that a more robust reduction of CRP after the first dose of the drug predicted a greater clinical benefit. The treatment was associated with a higher incidence of fatal infection than was placebo. An exploratory analysis showed a marked reduction in the incidence of lung cancer, as well as lung cancer mortality and total cancer mortality [53,54,55]. These results suggest that IL-1 is a marker of disease activity. Nevertheless, altering its levels might be of prognostic value. The lack of benefit of the 150-mg dose compared to other doses will be explored in future trials [56].
IL-1 in Arrhythmias
Investigations into the electrophysiological effects of IL-1β have centered on changes in Ca2+ handling and cell–cell coupling [4]. IL-1 was shown to significantly prolong action potential duration in guinea pig ventricular cells through changes in the conductance of calcium channels [57]. Recent studies showed that IL-1α increased the duration of the action potential in rat atrial myocardium, while IL-1β caused an appearance of extra-systolic patterns [58]. In a model of diabetic mice, IL-1β causes prolongation of the action potential duration, induces a decrease in potassium current and an increase in calcium sparks in cardiomyocytes, which are changes that underlie arrhythmia propensity. These arrhythmias can be successfully treated by inhibiting the IL-1β axis with either IL-1 receptor antagonist or by inhibiting the NLRP3 inflammasome [59]. Numerous other studies have found a decrease in L-type Ca2+ current by IL-1β [60]. Decreased responsiveness of L-type Ca2+ current to β-adrenergic stimulation has also been observed with IL-1β [15, 16]. IL-1β alone appears to have a significant impact on sarcoplasmic reticulum (SR) Ca2+ release and reuptake [61]. Several reports indicate that IL-1β decreases expression of important Ca2+ handling proteins, including ryanodine receptors (RyR), SR Ca2+ -ATPase (SERCA), and phospholamban (PLB) [16, 62, 63].
IL-1β was also shown to decrease connexin 43 (Cx43), a major protein in cardiac gap junctions that is believed to play a crucial role in the synchronized contraction of the heart. This was found in post-MI mouse and canine cardiac myocytes [64]. IL-1β produces cell–cell uncoupling, internalization, and reduced expression of Cx43. In an interesting model of atherosclerotic mice, post-MI hearts of atherosclerotic mice had a threefold increase in IL-1β expression, a twofold decrease in Cx43 expression, with a parallel increase in inducible ventricular arrhythmias. Furthermore, results could be reproduced with non-atherosclerotic post-MI mice that were exposed to lipopolysaccharide (LPS), which emphasizes the role of inflammation in deleterious electrophysiological consequences [65].
In a cohort of 112 RA patients, IL-1β, among other pro-inflammatory cytokines, was positively correlated with corrected QT (QTc) interval [66]. Patients with connective tissue diseases, positive anti-RO/SSA, and long QTc interval were found to have significantly higher levels of IL-1β [67]. A recent review suggested that the cause of prolonged QT in inflammatory conditions is cytokine induction of reactive oxygen species and ceramides [68].
In a case–control study of 122 patients with atrial fibrillation (AF) and 63 non-AF controls, IL-1 levels were significantly higher in AF patients compared to control patients. Patients with permanent and persistent AF had higher serum levels of IL-1 than those with paroxysmal AF [69].
DNA sampling from 70 proven lone AF patients and 70 healthy subjects showed that the presence of allele 2 of variable number of the tandem repeat (VNTR) polymorphism of IL-1 receptor antagonist gene may cause increased risk for lone AF, which is probably due to the inadequate limitation of inflammatory reactions [70].
IL-1 in Pericarditis and Inflammatory Cardiomyopathy
Pericarditis covers a diverse group of diseases with numerous etiologies. Usually, acute pericarditis is triggered by a viral infection and perpetuated by an autoinflammatory response. Usually, the disease is self-limiting in a matter of weeks [71]. Nevertheless, some patients suffer from recurrences, without a preceding viral illness, a fact that emphasizes the role of the autoinflammatory response [72]. Foreign viral antigens may share similar antigenic sequences and structures with the susceptible host’s antigens. Via molecular mimicry, the resulting cross-reactivity may lead to recurrent autoinflammatory episodes of pericarditis [73]. Regardless of the specific virus, dysfunctional response of the inflammasome is the pillar of the autoinflammatory response in the pericard [13].
In one case series, 13 adults with refractory pericarditis were treated with anakinra 100 mg/day subcutaneously. Most of them experienced clinical improvement within 2–5 days. Of all patients, only two still needed a low-dose corticosteroid treatment in a median follow-up of 23 months [74]. One more case series described ten adults suffering from idiopathic recurrent pericarditis who were treated with anakinra at a subcutaneous dose of 100 to 150 mg/day. The patients showed a prompt clinical improvement allowing steroid treatment cessation. However, seven patients suffered from recurrence following termination of anakinra [75]. More recently, the pilot AIRTRIP study was published, a randomized trial involving 21 patients with recurrent pericarditis at three Italian centers [76]. The patients had three or more episodes of pericarditis associated with an elevated CRP, were steroid dependent, and did not benefit from colchicine treatment. Anakinra was administered at 2 mg/kg per day up to 100 mg, for 2 months. Then, patients who responded with resolution of pericarditis were randomized to continuing anakinra (11 patients) or changing to placebo (ten patients) for 6 months, or until another recurrence of pericarditis. In a median follow-up of 14 months, Recurrent pericarditis occurred in nine of ten patients assigned to placebo, compared to two of 11 patients assigned to anakinra.
Acute myocarditis is an inflammatory disease of the myocardium that may be confirmed using imaging and/or histopathologic criteria. The spectrum of clinical manifestation is wide and ranges between mild dyspnea or chest pain that resolve spontaneously and lethal arrhythmias or cardiogenic shock. Different series attribute 10% of unexplained dilated cardiomyopathy (DCM) and 4–20% of sudden cardiac death (SCD) in young adults to myocarditis [6].
Recent studies indicate that IL-1 is central to the development of cardiac inflammation in the pathogenesis of a broad spectrum of conditions. Mechanistically, during myocardial injury, dying myocytes release various intracellular debris and mediators, including IL-1α, which strongly induce the innate immune response [77], activating the Toll-like receptors (TLRs), in particular TLR2 and TLR4, and the inflammasome [78]. Cardiac TLR4 mRNA expression has been found to be higher in patients with myocarditis than controls, and to correlate with viral RNA values in the heart [79]. IL1β is known to induce cardiac remodeling that leads to fibrosis, DCM, and HF following acute myocarditis in mice [80].
In fulminant myocarditis, the release of IL-1β results in widespread inflammation, leading to further death of cardiomyocytes, progressive loss of viable contractile tissue, and development of cardiomyopathy and heart failure [81].
In general, myocardial involvement is part of the clinical spectrum of inflammatory or autoinflammatory diseases, which are characteristically mediated by IL-1, such as adult-onset Still’s disease (AOSD). IL-1 receptor blockade was found highly effective in several case reports presenting patients with AOSD and secondary fulminant myocarditis, which was successfully treated with anakinra [82].
Furthermore, other case reports describe fulminant myocarditis patients, without a medical history of auto-inflammatory disease, which improved remarkably after initiating treatment with anakinra. Some of these patients recovered from cardiogenic shock requiring mechanical support including venous-arterial extracorporeal membrane oxygenation (ECMO) and mechanical circulatory support with a left ventricular assist device (LVAD) [83].
IL-1 in Sepsis-Induced Cardiomyopathy
In oppose to the early phase of septic shock that classically includes hyperdynamic left ventricle, low systemic vascular resistance and warm periphery (“warm shock”), the late phase comprises global left ventricular systolic dysfunction, poor peripheral perfusion, cool periphery (“cold shock”), and finally, death. Sepsis-induced cardiomyopathy (SIC) is a reversible myocardial dysfunction that typically resolves in 7–10 days. It is characterized by left ventricular dilatation and depressed ejection fraction [84, 85].
The pathophysiology of sepsis is believed to involve multifaceted interactions between pathogens and the immune system. The term “cytokine storm” relates to the breach of balance between the desirable inflammatory response and the activity of anti-inflammatory cytokines, with a corresponding tissue injury. Excess inflammatory mediators stimulate the production of reactive oxygen and nitrogen species, causing adjacent tissue injury with the further release of DAMPs that enhance the inflammatory process, creating a positive feedback circle that finally leads to cardiac dysfunction, multiple organ failure, and death [7].
While earlier theories suggested that global myocardial ischemia might be responsible for SIC, different studies suggested that it cannot explain this phenomenon [86]. An alternative concept, of a direct myocardial depression, is considered the major mechanism, with attenuation of the adrenergic response at the cardiomyocyte level due to down-regulation of β-adrenergic receptors and depression of post-receptor signaling pathways. Different mechanisms for myocardial depression involving IL-1 have been suggested. First, being a direct myocardial depressant factor [87]. Second, causing NO synthase expression in the myocardium, which increases total levels of sarcoplasmic reticulum Ca2+ and myocardial dysfunction [88]. Third, IL-1 might lead to mitochondrial dysfunction, with diminished activities of complexes I and II of the mitochondrial respiratory chain [18].
Treatment options rely on the basic approach to sepsis, as defined by early goal-directed therapy and surviving sepsis campaign bundles, with prompt and adequate antibiotic therapy, accompanied by surgical removal of the infectious focus, if indicated and feasible, while maintaining adequate arterial pressure using fluid resuscitation and norepinephrine. Other specific agents that were evaluated in the setup of SIC, such as dobutamine, levosimendan, milrinone, recombinant thrombomodulin, and beta-blockers, have not yet shown to positively affect clinical outcomes. Studies regarding direct IL-1 blockade in this setup are still lacking.
Conclusions
As we described, it seems that inflammation plays a role in the pathogenesis of many heart diseases, with IL-1 being one of the main mediators in this inflammatory process. Major cellular mechanisms involving IL-1 in heart diseases are summarized in Table 1. Until recently, animal models, observational data, and small pilot studies implied an encouraging effect of IL-1 blockade in coronary heart disease, heart failure, pericarditis, and myocarditis. Table 2 summarizes the current clinical trials evaluating IL-1 blockade. Initial small clinical trials have shown reduced CRP levels and improvement in cardiopulmonary exercise test parameters. Clinical trials in the setup of coronary disease have described better LV volume indices in STEMI patients treated with anakinra, with a reduction of new-onset HF. On the other hand, a larger clinical trial in NSTEMI patients showed a reduction in inflammatory markers in the anakinra group, but with no effect on infarct size on CMR or MACE at 30 and 90 days. The anakinra group even had higher rates of MACE in 12 months. The only randomized trial regarding patients with recurrent pericarditis was very small, but described impressive results when comparing anakinra to placebo. The recently published pivotal clinical trial of IL-1β blockade in the secondary prevention of patients with myocardial infarction (CANTOS) marks a new therapeutic target and might indicate a new era in cardiovascular medicine. Yet, while the beneficial effects described in the study are statistically significant, their clinical significance remains questionable. The ability of anti-inflammatory treatment in general, and IL-1 blockade in particular, to reduce morbidity and mortality in heart diseases, with a reasonable cost and adverse effects, still needs to be evaluated.
References
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–32. https://doi.org/10.1182/blood-2010-07-273417.
Braunwald E. Biomarkers in heart failure. N Engl J Med. 2008;358(20):2148–59. https://doi.org/10.1056/NEJMra0800239.
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–95. https://doi.org/10.1056/NEJMra043430.
Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol. 2016;91:114–22. https://doi.org/10.1016/j.yjmcc.2015.12.024.
Imazio M, Gaita F. Diagnosis and treatment of pericarditis. Heart Br Card Soc. 2015;101(14):1159–68. https://doi.org/10.1136/heartjnl-2014-306362.
Elamm C, Fairweather D, Cooper LT. Pathogenesis and diagnosis of myocarditis. Heart Br Card Soc. 2012;98(11):835–40. https://doi.org/10.1136/heartjnl-2012-301686.
Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22. https://doi.org/10.1186/s40560-016-0148-1.
Van Tassell BW, Raleigh JMV, Abbate A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep. 2015;12(1):33–41. https://doi.org/10.1007/s11897-014-0231-7.
Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128(17):1910–23. https://doi.org/10.1161/CIRCULATIONAHA.113.003199.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–147.
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–68. https://doi.org/10.1146/annurev.immunol.25.022106.141627.
Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3(105):cm1. https://doi.org/10.1126/scisignal.3105cm1.
Cremer PC, Kumar A, Kontzias A, Tan CD, Rodriguez ER, Imazio M, Klein AL. Complicated pericarditis: understanding risk factors and pathophysiology to inform imaging and treatment. J Am Coll Cardiol. 2016;68(21):2311–28. https://doi.org/10.1016/j.jacc.2016.07.785.
Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Systemic inflammation in heart failure–the whys and wherefores. Heart Fail Rev. 2006;11(1):83–92. https://doi.org/10.1007/s10741-006-9196-2.
Liu SJ, Zhou W, Kennedy RH. Suppression of beta-adrenergic responsiveness of L-type Ca2+ current by IL-1beta in rat ventricular myocytes. Am J Physiol. 1999;276(1 Pt 2):H141–8.
Combes A, Frye CS, Lemster BH, Brooks SS, Watkins SC, Feldman AM, McTiernan CF. Chronic exposure to interleukin 1beta induces a delayed and reversible alteration in excitation-contraction coupling of cultured cardiomyocytes. Pflugers Arch. 2002;445(2):246–56. https://doi.org/10.1007/s00424-002-0921-y.
Tatsumi T, Matoba S, Kawahara A, Keira N, Shiraishi J, Akashi K, Kobara M, Tanaka T, Katamura M, Nakagawa C, Ohta B, Shirayama T, Takeda K, Asayama J, Fliss H, Nakagawa M. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs contractile function in rat cardiac myocytes. J Am Coll Cardiol. 2000;35(5):1338–46.
Zell R, Geck P, Werdan K, Boekstegers P. Tnf-α and IL-1α inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol Cell Biochem. 1997;177(1):61–7. https://doi.org/10.1023/A:1006896832582.
Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, Lee RT. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. 2009;2(6):684–91. https://doi.org/10.1161/CIRCHEARTFAILURE.109.873240.
Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the Valsartan Heart Failure Trial. Circ Heart Fail. 2014;7(3):418–26. https://doi.org/10.1161/CIRCHEARTFAILURE.113.001036.
Felker GM, Fiuzat M, Thompson V, Shaw LK, Neely ML, Adams KF, Whellan DJ, Donahue MP, Ahmad T, Kitzman DW, Pina IL, Zannad F, Kraus WE, O’Connor CM. Soluble ST2 in ambulatory patients with heart failure: association with functional capacity and long-term outcomes. Circ Heart Fail. 2013;6(6):1172–9. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000207.
Daniels LB, Bayes-Genis A. Using ST2 in cardiovascular patients: a review. Future Cardiol. 2014;10(4):525–39. https://doi.org/10.2217/fca.14.36.
Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res Off J Eur Histamine Res Soc Al. 2013;62(7):637–40. https://doi.org/10.1007/s00011-013-0625-0.
Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, Shah K, Canada J, Voelkel NF, Dinarello CA, Abbate A. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 2012;7(3):e33438. https://doi.org/10.1371/journal.pone.0033438.
Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183(3):949–58.
Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, Zhu S, Zhang R, Han W. Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur J Pharmacol. 2010;643(2–3):247–53. https://doi.org/10.1016/j.ejphar.2010.06.024.
Mezzaroma E, Mikkelsen RB, Toldo S, Mauro AG, Sharma K, Marchetti C, Alam A, Van Tassell BW, Gewirtz DA, Abbate A. Role of Interleukin-1 in Radiation-Induced Cardiomyopathy. Mol Med Camb Mass. 2015;21:210–8. https://doi.org/10.2119/molmed.2014.00243.
Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, Katsimbri P, Skarantavos G, Soucacos PN, Kremastinos DT. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117(20):2662–9. https://doi.org/10.1161/CIRCULATIONAHA.107.731877.
Abbate A, Canada JM, Van Tassell BW, Wise CM, Dinarello CA. Interleukin-1 blockade in rheumatoid arthritis and heart failure: a missed opportunity? Int J Cardiol. 2014;171(3):e125–6. https://doi.org/10.1016/j.ijcard.2013.12.078.
Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, Turlington JS, Thurber CJ, Christopher S, Dixon DL, Fronk DT, Thomas CS, Rose SW, Buckley LF, Dinarello CA, Biondi-Zoccai G, Abbate A. Interleukin-1 blockade in acute decompensated heart failure: a randomized, double-blinded, Placebo-Controlled Pilot Study. J Cardiovasc Pharmacol. 2016;67(6):544–51. https://doi.org/10.1097/FJC.0000000000000378.
Van Tassell BW, Arena R, Biondi-Zoccai G, McNair Canada J, Oddi C, Abouzaki NA, Jahangiri A, Falcao RA, Kontos MC, Shah KB, Voelkel NF, Dinarello CA, Abbate A. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. 2014;113(2):321–7. https://doi.org/10.1016/j.amjcard.2013.08.047.
Van Tassell BW, Buckley LF, Carbone S, Trankle CR, Canada JM, Dixon DL, Abouzaki N, Oddi-Erdle C, Biondi-Zoccai G, Arena R, Abbate A. Interleukin-1 blockade in heart failure with preserved ejection fraction: rationale and design of the diastolic heart failure anakinra response trial 2 (D-HART2). Clin Cardiol. 2017. https://doi.org/10.1002/clc.22719.
Saxena A, Russo I, Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res J Lab Clin Med. 2016;167(1):152–66. https://doi.org/10.1016/j.trsl.2015.07.002.
Lugrin J, Parapanov R, Rosenblatt-Velin N, Rignault-Clerc S, Feihl F, Waeber B, Muller O, Vergely C, Zeller M, Tardivel A, Schneider P, Pacher P, Liaudet L. Cutting edge: IL-1alpha is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction. J Immunol. 2015;194(2):499–503. https://doi.org/10.4049/jimmunol.1401948.
Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, Frangogiannis NG. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164(2):665–77. https://doi.org/10.1016/S0002-9440(10)63154-9.
Christia P, Bujak M, Gonzalez-Quesada C, Chen W, Dobaczewski M, Reddy A, Frangogiannis NG. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem Off J Histochem Soc. 2013;61(8):555–70. https://doi.org/10.1369/0022155413493912.
Libby P, Warner SJ, Friedman GB. Interleukin 1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J Clin Invest. 1988;81(2):487–98. https://doi.org/10.1172/JCI113346.
Offner FA, Feichtinger H, Stadlmann S, Obrist P, Marth C, Klingler P, Grage B, Schmahl M, Knabbe C. Transforming growth factor-beta synthesis by human peritoneal mesothelial cells. Induction by interleukin-1. Am J Pathol. 1996;148(5):1679–88.
Cozzolino F, Torcia M, Aldinucci D, Ziche M, Almerigogna F, Bani D, Stern DM. Interleukin 1 is an autocrine regulator of human endothelial cell growth. Proc Natl Acad Sci USA. 1990;87(17):6487–91.
Bochner BS, Luscinskas FW, Gimbrone MAJ, Newman W, Sterbinsky SA, Derse-Anthony CP, Klunk D, Schleimer RP. Adhesion of human basophils, eosinophils, and neutrophils to interleukin. J Exp Med. 1991;173(6):1553–7.
Grames M, Breviario F, Pintucci G, Millet I, Dejana E, van Damme J, Donati MB, Mussoni L. Enhancement by interleukin-1 (IL-1) of plasminogen activator inhibitor (PA-I) activity in cultured human endothelial cells. Biochem Biophys Res Commun. 1986;139(2):720–7.
Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V, Chamberlain J, Francis S. Neutrophil elastase promotes interleukin-1beta secretion from human coronary endothelium. J Biol Chem. 2015;290(40):24067–78. https://doi.org/10.1074/jbc.M115.659029.
Chai M, Zhang H-T, Zhou Y-J, Ji Q-W, Yang Q, Liu Y-Y, Zhao Y-X, Shi D-M, Liu W, Yang L-X, Zhang L-L, Liang J. Elevated IL-37 levels in the plasma of patients with severe coronary artery calcification. J Geriatr Cardiol JGC. 2017;14(5):285–91. https://doi.org/10.11909/j.issn.1671-5411.2017.05.013.
Liu Z, Zhang M, Wu J, Zhou P, Liu Y, Wu Y, Yang Y, Lu X. Serum CD121a (Interleukin 1 Receptor, Type I): a potential novel inflammatory marker for coronary heart disease. PLoS One. 2015;10(6):e0131086. https://doi.org/10.1371/journal.pone.0131086.
Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173(1):57–67. https://doi.org/10.2353/ajpath.2008.070974.
Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GGL, Houser J-E, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117(20):2670–83. https://doi.org/10.1161/CIRCULATIONAHA.107.740233.
Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, Abbate A. Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. 2013;98(3):734–45. https://doi.org/10.1113/expphysiol.2012.069831.
Ikonomidis I, Tzortzis S, Andreadou I, Paraskevaidis I, Katseli C, Katsimbri P, Pavlidis G, Parissis J, Kremastinos D, Anastasiou-Nana M, Lekakis J. Increased benefit of interleukin-1 inhibition on vascular function, myocardial deformation, and twisting in patients with coronary artery disease and coexisting rheumatoid arthritis. Circ Cardiovasc Imaging. 2014;7(4):619–28. https://doi.org/10.1161/CIRCIMAGING.113.001193.
Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol. 2010;105(10):1371–7. https://doi.org/10.1016/j.amjcard.2009.12.059.
Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol. 2013;111(10):1394–400. https://doi.org/10.1016/j.amjcard.2013.01.287.
Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW, Oddi C, Carbone S, Trankle CR, Roberts CS, Mueller GH, Gambill ML, Christopher S, Markley R, Vetrovec GW, Dinarello CA, Biondi-Zoccai G. Comparative safety of interleukin-1 blockade with anakinra in patients with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am J Cardiol. 2015;115(3):288–92. https://doi.org/10.1016/j.amjcard.2014.11.003.
Morton AC, Rothman AMK, Greenwood JP, Gunn J, Chase A, Clarke B, Hall AS, Fox K, Foley C, Banya W, Wang D, Flather MD, Crossman DC. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J. 2015;36(6):377–84. https://doi.org/10.1093/eurheartj/ehu272.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017. https://doi.org/10.1056/NEJMoa1707914.
Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet Lond Engl. 2017;390(10105):1833–42. https://doi.org/10.1016/S0140-6736(17)32247-X.
Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet Lond Engl. 2017. https://doi.org/10.1016/S0140-6736(17)32814-3.
Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125(7):605–13.
Li YH, Rozanski GJ. Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc Res. 1993;27(3):525–30.
Mitrokhin VM, Mladenov MI, Kamkin AG. IL-1 provokes electrical abnormalities in rat atrial myocardium. Int Immunopharmacol. 2015;28(1):780–4. https://doi.org/10.1016/j.intimp.2015.08.006.
Monnerat G, Alarcon ML, Vasconcellos LR, Hochman-Mendez C, Brasil G, Bassani RA, Casis O, Malan D, Travassos LH, Sepulveda M, Burgos JI, Vila-Petroff M, Dutra FF, Bozza MT, Paiva CN, Carvalho AB, Bonomo A, Fleischmann BK, de Carvalho ACC, Medei E. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7:13344. https://doi.org/10.1038/ncomms13344.
El Khoury N, Mathieu S, Fiset C. Interleukin-1beta reduces L-type Ca2+ current through protein kinase C activation in mouse heart. J Biol Chem. 2014;289(32):21896–908. https://doi.org/10.1074/jbc.M114.549642.
Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47(4):378–86. https://doi.org/10.1016/j.ceca.2010.02.002.
Patten M, Hartogensis WE, Long CS. Interleukin-1beta is a negative transcriptional regulator of alpha1-adrenergic induced gene expression in cultured cardiac myocytes. J Biol Chem. 1996;271(35):21134–41.
McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81(4):493–503.
Baum JR, Long B, Cabo C, Duffy HS. Myofibroblasts cause heterogeneous Cx43 reduction and are unlikely to be coupled to myocytes in the healing canine infarct. Am J Physiol Heart Circ Physiol. 2012;302(3):H790–800. https://doi.org/10.1152/ajpheart.00498.2011.
De Jesus NM, Wang L, Herren AW, Wang J, Shenasa F, Bers DM, Lindsey ML, Ripplinger CM. Atherosclerosis exacerbates arrhythmia following myocardial infarction: role of myocardial inflammation. Heart Rhythm. 2015;12(1):169–78. https://doi.org/10.1016/j.hrthm.2014.10.007.
Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol. 2015;42(3):421–8. https://doi.org/10.3899/jrheum.140861.
Pisoni CN, Reina S, Arakaki D, Eimon A, Carrizo C, Borda E. Elevated IL-1beta levels in anti-Ro/SSA connective tissue diseases patients with prolonged corrected QTc interval. Clin Exp Rheumatol. 2015;33(5):715–20.
Sordillo PP, Sordillo DC, Helson L. Review: the prolonged QT interval: role of pro-inflammatory cytokines, reactive oxygen species and the ceramide and sphingosine-1 phosphate pathways. Vivo Athens Greece. 2015;29(6):619–36.
Cheng T, Wang X-F, Hou Y-T, Zhang L. Correlation between atrial fibrillation, serum amyloid protein A and other inflammatory cytokines. Mol Med Rep. 2012;6(3):581–4. https://doi.org/10.3892/mmr.2012.934.
Gungor B, Ekmekci A, Arman A, Ozcan KS, Ucer E, Alper AT, Calik N, Yilmaz H, Tezel T, Coker A, Bolca O. Assessment of interleukin-1 gene cluster polymorphisms in lone atrial fibrillation: new insight into the role of inflammation in atrial fibrillation. Pacing Clin Electrophysiol PACE. 2013;36(10):1220–7. https://doi.org/10.1111/pace.12182.
Xu B, Harb SC, Cremer PC. New insights into pericarditis: mechanisms of injury and therapeutic targets. Curr Cardiol Rep. 2017;19(7):60. https://doi.org/10.1007/s11886-017-0866-6.
Imazio M. Idiopathic recurrent pericarditis as an immune-mediated disease: current insights into pathogenesis and emerging treatment options. Expert Rev Clin Immunol. 2014;10(11):1487–92. https://doi.org/10.1586/1744666X.2014.965150.
Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol. 2012;42(1):102–11. https://doi.org/10.1007/s12016-011-8294-7.
Jain S, Thongprayoon C, Espinosa RE, Hayes SN, Klarich KW, Cooper LT, Moder KG, Anavekar NS, Oh JK, Matteson EL. Effectiveness and safety of anakinra for management of refractory pericarditis. Am J Cardiol. 2015;116(8):1277–9. https://doi.org/10.1016/j.amjcard.2015.07.047.
Lazaros G, Vasileiou P, Koutsianas C, Antonatou K, Stefanadis C, Pectasides D, Vassilopoulos D. Anakinra for the management of resistant idiopathic recurrent pericarditis. Initial experience in 10 adult cases. Ann Rheum Dis. 2014;73(12):2215–7. https://doi.org/10.1136/annrheumdis-2014-205990.
Brucato A, Imazio M, Gattorno M, Lazaros G, Maestroni S, Carraro M, Finetti M, Cumetti D, Carobbio A, Ruperto N, Marcolongo R, Lorini M, Rimini A, Valenti A, Erre GL, Sormani MP, Belli R, Gaita F, Martini A. Effect of anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: the AIRTRIP randomized clinical trial. JAMA. 2016;316(18):1906–12. https://doi.org/10.1001/jama.2016.15826.
Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, Azam T, Kim S, Dinarello CA. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol. 2013;4:391. https://doi.org/10.3389/fimmu.2013.00391.
Toldo S, Kannan H, Bussani R, Anzini M, Sonnino C, Sinagra G, Merlo M, Mezzaroma E, De-Giorgio F, Silvestri F, Van Tassell BW, Baldi A, Abbate A. Formation of the inflammasome in acute myocarditis. Int J Cardiol. 2014;171(3):e119–21. https://doi.org/10.1016/j.ijcard.2013.12.137.
Satoh M, Nakamura M, Akatsu T, Iwasaka J, Shimoda Y, Segawa I, Hiramori K. Expression of Toll-like receptor 4 is associated with enteroviral replication in human myocarditis. Clin Sci Lond Engl. 2003;104(6):577–84. https://doi.org/10.1042/cs20020263.
Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, Pedrazzini T, Berger CT, Dirnhofer S, Matter CM, Penninger JM, Luscher TF, Eriksson U. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. 2009;105(9):912–20. https://doi.org/10.1161/CIRCRESAHA.109.199802.
Gupta S, Markham DW, Drazner MH, Mammen PP. Fulminant myocarditis. Nat Clin Pract Cardiovasc Med. 2008;5(11):693–706. https://doi.org/10.1038/ncpcardio1331.
Parisi F, Paglionico A, Varriano V, Ferraccioli G, Gremese E. Refractory adult-onset Still disease complicated by macrophage activation syndrome and acute myocarditis: a case report treated with high doses (8 mg/kg/d) of anakinra. Medicine (Baltimore). 2017;96(24):e6656. https://doi.org/10.1097/MD.0000000000006656.
Cavalli G, Foppoli M, Cabrini L, Dinarello CA, Tresoldi M, Dagna L. Interleukin-1 receptor blockade rescues myocarditis-associated end-stage heart failure. Front Immunol. 2017;8:131. https://doi.org/10.3389/fimmu.2017.00131.
Sato R, Nasu M. A review of sepsis-induced cardiomyopathy. J Intensive Care. 2015;3:48. https://doi.org/10.1186/s40560-015-0112-5.
Papaioannou VE (2015) Frontiers in myocardia: septic cardiomyopathy from bench-to-bedside. Bentham Science Publishers. https://books.google.co.il/books?id=yfn8DQAAQBAJ
Cunnion RE, Schaer GL, Parker MM, Natanson C, Parrillo JE. The coronary circulation in human septic shock. Circulation. 1986;73(4):637–44.
Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183(3):949–58.
Kumar A, Brar R, Wang P, Dee L, Skorupa G, Khadour F, Schulz R, Parrillo JE. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276(1 Pt 2):R265–76.
Acknowledgements
Funding
No funding or sponsorship was received for this study or for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Yishay Szekely and Yaron Arbel have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to https://doi.org/10.6084/m9.figshare.5811330.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szekely, Y., Arbel, Y. A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today?. Cardiol Ther 7, 25–44 (2018). https://doi.org/10.1007/s40119-018-0104-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-018-0104-3