
Abstract
In situ nanoparticle formation embedded into hydrogel matrix, acting as container and stabilizer for nanoparticle reaction was the focus in this research; this method was realized using AgNO3 (0.75 and 1.0 M) as silver source for nanoparticle formation; also, monomers (HEMA), cross-linker agents (DEGDMA) and a photoinitiator (Irgacure 651) were used for the hydrogel synthesis. For the reduction of Ag+ → Ag0, the reaction mixture was irradiated with an UV lamp at 365 nm for 30 min; parallel to this process, the hydrogel photopolymerization occurs. All these systems were studied by Infrared and Raman Spectroscopy, optical studies: UV/Vis absorption, thermal studies: differential scanning calorimetry and thermogravimetric analysis, X-ray diffraction, fluorescence X-ray spectroscopy and transmission electronic microscopy. Characterization techniques are capable to detect the presence of non-agglomerated silver nanoparticles homogeneously distributed in all the systems. X-Ray photoemission spectroscopy establishes the presence of Ag0 and Ag+ as mixture in the synthesized composite. Quantitative assays show that the sample Ag_Hg3-89.5 % (1.0 M) presents an important biocide property, by reducing 99.9 % of bacterium Escherichia coli ATCC 25922 as compared with the alone hydrogel used as control.
GRAPHICAL ABSTRACT
The embedding of silver nanoparticles homogenously distributed and stabilized into the hydrogel matrix was realized in situ using a photoinitiator during the synthesis. In figures it is possible to observe different conformations of hydrogel/silver nanoparticle composites, so the non-organic nanoparticles seem to be confined in the organic polymer matrix, forming a bunch. TEM analysis shows spherical nanoparticles with diameters around 22–37 nm. Biocide tests show that this compound could be utilized as E. coli bactericide with remarkable results.

Similar content being viewed by others

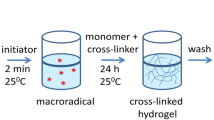
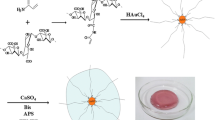
Introduction
In the last 15 years, the research related to structured and intelligent materials with specified nanoproperties has had an incredible and remarkable increase. This is related to the wide range of applications that offer these kinds of materials. In this context, intelligent hydrogels have attracted the attention of several researchers due to their hydrophilic character and their potential biocompatibilism [1–4].
Since the first reported research on crosslinked 2-hydroxyethyl methacrylate (HEMA)-based hydrogels by Wichterle et al. [5], these types of polymers have become quite interesting due to their characteristics and the multiple kinds of applications. These applications have been increasing from those days to date [6–13]. Hydrogel crosslinking networks are capable to absorb from 10 to 20 % to thousands times their dry weight in water maintaining their three-dimensional network structure; they are chemically stable in some conditions and also could be degraded and eventually dissolved in other ones. Another important fact is that the swelling process is reversible. Hydrogels based on both natural and synthetic polymers still are of interest for cell encapsulation applications [14–17]. Recently, such hydrogels have become especially attractive for the field of tissue engineering as matrices for repairing and regenerating a wide variety of tissues and organs [18–21]; also these materials are actually used for the fabrication of drug delivery systems, separation operations in biotechnology, processing of agricultural products, conductive and superabsorbent composites, sensors and actuators [22–24]. These materials exhibit a swelling response to environmental changes such as temperature, pH, electric fields, UV or visible light radiation, among others.
Hydrogels may be synthesized via several different ways such as thermal- [25], redox- [26], RAFT- [27, 28] and photopolymerization [29]. This last method that corresponds to a photoinitiated radical polymerization of multifunctional monomers that produce highly crosslinked polymers, has several advantages over conventional ones like spatial and temporal control, fast curing and reaction rates at room temperature, solvent-free formulation, among others. The photopolymerization process has a unique reaction behavior that includes unequal functional group reactivity, self-acceleration and self-deceleration induced by diffusion-controlled propagation and termination, among others [30, 31]. Resulting hydrogels in which HEMA was used as monomer precursor show a high surface porosity and swelling degree compared to other ones, being a good candidate for drug delivery and others applications [32–34].
Many comonomers are added to adjust the crosslinking density, swelling and mechanical properties of the hydrogels. For instance, diethylene glycol dimethacylate (DEGDMA) is usually mixed with HEMA for preparing poly(HEMA) hydrogels to enhance the reaction kinetics and physical hydrogel properties [35]. In Fig. 1, two- and three-dimensional chemical structures of HEMA and DEGDMA are shown, respectively.

HEMA and DEGDMA structures
Nanosized inorganic particles display unique physical and chemical properties and represent an increasingly important field in the development of nanodevices . These last ones could be used in numerous biomedical, catalytical, optical, electronic and quantum-size domain applications [36–39]. In particular, silver nanoparticles have recently been used as antimicrobial agent for some Gram-negative bacteria [40–44]. The focus of this work is to take advantage of this latter feature and to combine some hydrogel applications, such as drug delivery, with the antimicrobial effect; for these reasons, it has developed a novel method for synthesizing silver nanoparticles while hydrogel is forming and stabilize them through free radical reaction with the mixture during the hydrogel photopolymerization.
Silver nanoparticles had demonstrated antimicrobial activity at the level of DNA damage, cytoplasmic membrane, etc. These behaviors were studied by Kim et al., showing that Ag–NPs have a powerful antibacterial activity against S. aureus cells and E. coli; increases in cell membrane permeability can be evidence for the formation of Ag–NPs “pits” on the cell walls. Likewise, reduced lactate dehydrogenase (LDH) activity also can be consequence of reactive oxygen species (ROS) formed by Ag–NPs [45].
Several studies have shown that the ROS could cause damage to the plasma membrane, structure of proteins, DNA and bacterial respiratory chain. The thick peptidoglycan layer of Gram-positive bacteria may be better protection against pits or ROS formed by Ag–NPs than thin peptidoglycan layer of Gram-negative bacteria as E. coli. Similar data using silver nanoparticles in hydrogels have shown good biocide activity against E. coli. Varsha et al. [46] showed a 78.45 % reduction percentage of E. coli bacterial while were treated with nanosilver-loaded chitosan attached for 24 h. Moreover, the antibacterial action of hydrogels is a determining factor for applications in water disinfection devices. Agnihotri et al. [47] tested nano Ag–Ag-loaded hydrogels against E. coli MTCC443 strain; they showed a reduction of 83.5 % in E. coli growth after 12 h.
A complete system characterization was realized by spectroscopic methods such as Infrared Spectroscopy (FT-IR), Raman Spectroscopy and UV/Vis spectroscopy. The first two techniques corroborate the chemical structure via the identification of some polymer characteristic groups, and the last one was performed to verify the presence of silver nanoparticles into the system. Also, differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were carried out with the finality to learn more about their thermal behavior. X-ray diffraction was realized to find some type of crystalline ordering due to the presence of silver nanoparticles into the system. Fluorescence X-ray Spectroscopy (EDX-XRF) was used to measure the quantity of silver nanoparticles embedded into the hydrogel matrix. Together with these studies, X-ray photoemission spectroscopy (XPS) technique was used to determine the silver oxidation state into the hydrogel. Moreover, micrographs with transmission electronic microscopy (TEM) were taken to characterize the structure, size and spatial dispersion of the silver nanoparticles. Finally, antimicrobial behavior against E. coli of these hydrogels, with and without silver nanoparticles, was tested.
Experimental
Materials
HEMA (2-hydroxyethyl methacrylate, 97 %), containing ≤250 ppm monomethyl ether hydroquinone as inhibitor and DEGDMA (Di (ethylene glycol) dimethacrylate, 95 %), was used as crosslinking agent; Irgacure 651 (2,2-Dimethoxy-2-phenylacetophenone) was used as photoinitiator. All the reactants were purchased from Sigma Aldrich (St. Louis, MO, USA). Ultrapure water for chromatography LiChrosolv (Merck KGaA, Darmstadt, Germany) was used as dissolvent during the photopolymerization. In addition, hydrogel polymerizations were realized through sample radiation exposure using a UV lamp with the emission peak centered at 365 nm (VILBER LOURMAT 230 V 50/60 Hz, 365 nm, 9 W). Then, to eliminate all the water molecules occluded, the samples were subjected to a freeze-dried lyophilization process (Labconco FreeZone 6, 12 Lt., console freeze dry system with purge valve and mini chamber).
Equipments and measurements
FT-IR spectra (KBr pellet) were obtained on a Bruker TENSOR 27 (Bruker Optics Inc., Ettlingen, Germany) equipped with a KBr beamsplitter, RockSolid™ interferometer, DigiTect™ detector system and a 1.2-mW, 632.8 He–Ne laser. The structural and vibrational properties of the hydrogels and their composites (hydrogel–silver nanoparticles) were characterized by Raman spectroscopy with a LabRam 010 instrument from ISA equipped with a 5.5-mW, 632.8 nm He–Ne laser without filter. The Raman microscope uses a back-scattering geometry, where the incident beam is linearly polarized at 500:1 ratio. The microscope objective used to realize the Raman analysis was an Olympus Mplan 50x.
Absorption spectra of the hydrogels pure and their composites were measured in solid state using barium sulfate as standard. These results were recorded at 25 °C between 200 and 800 nm in a UV–Vis spectrophotometer Perkin-Elmer Lambda 35.
DSC traces were recorded on a Mettler Toledo, DSC 822e Differential Scanning Calorimetric from the second heating run. The thermal degradation studies were developed in a TGA/DSC 1 Star e System Mettler Toledo thermobalance. Both analyses were recorded with a heating rate of 20 °C min−1 under nitrogen atmosphere.
X-ray diffraction analysis were developed at room temperature with a Bruker D-8 Advanced Diffractometer, using a long, fine-focus ceramic X-ray tube with copper anode (λ(CuKα) = 1.54 Å) working at 2.2 kW. The diffraction patterns were obtained in the usual Bragg–Brentano (θ − 2θ) geometry via powder samples in a rotating holder. The X-ray tube was operated at 40 kV and 40 mA. The goniometer was swept between 5° and 140° with 0.02° steps in the whole interval. The diffracted X-rays were detected with a NaI dynamic scintillation detector.
Fluorescence X-ray Spectroscopy (EDX-XRF) was realized with a Shimadzu EDX-720 in vacuum through powder sample. The energy dispersion of the X-ray fluorescence was detected with a Si detector with liquid nitrogen for cooling. The X-ray was generated with an Rh anode with voltages between 5–50 kV and a current of 1–1,000 μA.
Chemical characterization was performed by XPS with a STAIB RQ300 X-ray source using Mg K-alpha x-radiation and a STAIB DESA100 electron spectrometer, which was operated in a constant pass energy mode at 225 W. The working pressure in the analysis chamber was typically 5 × 10−9 torr. The binding energy scale was calibrated by measuring the C 1 s peak at 285.0 eV. The composition and chemical state were investigated based on the areas and binding energies of O 1 s, C 1 s and Ag 3d photoelectron peaks. Data analysis was performed by a fitting program using properly mixed Lorentzian–Gaussian functions after background subtraction according to the Shirley method. Surface atomic compositions were determined using standard XPS cross sections.
The size and morphology of the nanoparticles were studied by TEM, model Philips Tecnai 12 BioTWIN operated at 120 kV with different magnifications. The grids were brought from Ted Pella, Inc.; 300 mesh copper specimen grids with Formvar/Carbon support films.
Quantitative assay of biocide properties
The antimicrobial effect of the hydrogel/silver nanoparticle compounds was determined using a plate count method. E. coli ATCC 25922 was used for analysis.
Quantitative assays were realized by bacterial killing determination using viable bacteria count through pour plate culture method. Both, hydrogel/silver nanoparticle and hydrogel alone (control samples) with 314 mm2 surface area, were suspended in 20 mL trypticase soy broth flask inoculated with an Escherichia coli ATCC 25922 culture with density of ~105 colony-forming units (CFU) mL−1, measured by Densimat bioMérieuxTM and incubated at 37 °C during 18 h at atmospheric ambient.
Trypticase soy aliquots of 500 μL from hydrogel/silver nanoparticle and hydrogel alone flasks were removed and serially diluted (1/10, 1/100, 1/1,000, 1/10,000 and 1/100,000); 0.2 mL of each dilution was plated in duplicate on trypticase soy agar plates and incubated at 37 °C for 24 h. After incubation, the number of colonies on the Petri dishes was counted, and the microorganism inhibition percentage in each sample was determined according to their respective control. Total bacterial CFU mL−1 (log 10 CFU mL−1) was determined after 24-h. incubation. The measures were realized using triplicate samples to obtain a dependable result.
The microorganism reduction percentage of each sample was determined according to its respective control using the following equation:
Results and discussion
Preparation of the hydrogels with the silver nanoparticles
The polymerization was carried out in transparent flasks: 2 g of HEMA (15.368 mmol), a variable amount of DEGDMA (for example, 416 mg over 1.717 mmol for Ag Hg3-89.5 % sample) and 22.00 mg of Irgacure 651 (0.0858 mmol) were added and dissolved in 1 mL of chromatography water. The reaction mixture was continuously purged with nitrogen gas to displace the air of the system avoiding the oxidation. Moreover, AgNO3 is poured (0.75 M and 1 M) into the mixture, keeping the oxygen-free environment, and then the solution was stirred for 30 min (in dark) to reach a complete homogenization. Afterwards, the flask was irradiated at 365 nm with a UV lamp for 30 min; the solution changes its color from transparent to light brown during the photopolymerization. The compounds were designated as: Ag Hg3-89.5; Ag Hg3-94.5; Ag Hg3-97.0; Ag Hg3-98.0 and Ag Hg3-98.5 %, these numbers are related to the following molar ratio between HEMA/DEGDMA: 15.368/1.717; 15.368/0.813; 15.368/0.396; 15.368/0.235; 15.368/0.1559, respectively. Once the hydrogels with silver nanoparticles were obtained, the samples were washed with distillated water to eliminate the precursors. Finally, the compounds were lyophilized for 48 h and then pulverized in an agatha mortar for finally obtain a powder sample that makes more suitable the characterization.
An aqueous solution of AgNO3 salt was used as source of Ag+ ions, due to its UV photosensitivity. AgNO3 is able to produce stable silver nanoparticles (Ag0); this process is accompanied with a color change (colorless → light brown) indicating formation of AgNPs, which is associated with a surface plasmon vibration of metal nanoparticles. These properties had been studied with UV–Vis spectroscopy, and the results that are discussed in optical characterization section. In addition, the formation of stable silver nanoparticle requires free radicals provided by the photoinitiator breaking (Irgacure 651) started with UV irradiation; this process competes directly against the polymerization because it requires those free radicals too.
FT-IR and Raman spectra analysis
Spectroscopies studies were realized to corroborate the chemical structure of the hydrogels and their composites via the identification of some characteristic groups. A summary of these studies is presented in next paragraph.
Hydrogel pure, Hg3-89.5 %
FT-IR, KBr, cm−1: 3,446 (stretching, O–H (free) very broad); 2,998–2,876 (stretching, CH3, CH2, CH three bands; OH (H-bonded) and possibly C–H of the aromatic group, m); 1,725 (stretching, C=O ester, m); 1,651–1,619 (stretching C=C ring, three possible bands, m); 1,460 (bending, CH2 and CH3 deformation, w); 1,387 (bending, C–O–H and CH3 deformation, w); 1,163 (stretching, C–O–OR ester, m); 1,069 (stretching C–OH, m); 845 (CH bending and ring puckering, w); 748 (bending CH2 rocking and OH bend (out-of-plane), w). Raman, cm−1: 3,470 (stretching, Hydrogen bonded OH intermolecular polymeric, s); 3,001–2,886 (stretching, CH3 (antisym.) and CH2 (sym.) vibration and C–H of the substituted benzenes, vs); 1,724 (stretching, unconjugated C=O, m); 1,596 (stretching C=C ring bands, vw); 1,455 (Bending, CH3 vibration, antisym., and CH2 scissoring, s); 1,389 (bending, involving –C–OH, w); 1,276–1,206, 1,121–1,087 (stretching, C–C skelt., s–m); 1,022 (stretching, out-of-phase C–C–OH, m); 967 (stretching C–O–R and bending, in-phase C–H for mono-substituted benzenes, m); 895–829 (stretching, C–C skelt. and in-phase C–C–OH, s–m) and 600 (bending, O–C=O in-plane deformation and in-plane C–H of ring, m).
Composite, Ag_Hg3-89.5 % (0.75 M)
FT-IR, KBr, cm−1: 3,440 (stretching, O–H (free) very broad); 2,948–2,872 (stretching, CH3, CH2, CH three bands; OH (H-bonded) and possibly C–H of the aromatic group, m); 1,716 (stretching, C=O ester, s); 1,655–1,629 (stretching C=C ring, three possible bands, m); 1,461 (bending, CH2 and CH3 deformation, m); 1,385 (bending, C–O–H and CH3 deformation, m); 1,171 (stretching, C–O–O ester, m); 1,078 (stretching C–OH, m); 847 (CH bending and ring puckering, w); 745 (bending CH2 rocking and OH bend (out-of-plane), w). Raman, cm−1: 3,479 (stretching, Hydrogen bonded OH intermolecular polymeric, s); 3,001–2,886 (stretching, CH3 (antisym.) and CH2 (sym.) vibration and C–H of the substituted benzenes, vs); 1,717 (stretching, unconjugated C=O, m); 1,598 (stretching C=C ring bands, vw); 1,452 (Bending, CH3 vibration, antisym., and CH2 scissoring, s); 1,391 (bending, involving –C–OH, w); 1,275–1,201, 1,122–1,081 (stretching, C–C skelt., s–m); 1,026 (stretching, out-of-phase C–C–OH, m); 967 (stretching C–O–R and bending, in-phase C–H for mono-substituted benzenes, m); 895–826 (stretching, C–C skelt. and in-phase C–C–OH, s–m) and 605 (bending, O–C=O in-plane deformation and in-plane C–H of ring, m).
According to the results obtained from FT-IR, the three bands shown in Fig. 2a, b could be related to the formation of the methyl benzoate produced by the fragmentation of the Irgacure 651, which is used as photoinitiator [48]. These bands correspond to stretching of the C–H and C=C aromatic group, bending CH and ring puckering. These results are based on the fact that the methyl benzoate is insoluble in water, assuming that the hydrogel cleaning do not affect the reactive elimination. This bands C=C and C–H also were corroborated by Raman spectroscopy.

FT-IR/Raman spectrum of: a hydrogel pure, Hg3-89.5 % and b Composite Ag_Hg3-89.5 % (0.75 M)
On the other hand, the medium-strong band related to the stretching vinyl group near 1,650–1,638 cm−1 is not observed by Raman [49]. FT-IR at 1,900–2,000 cm−1 is not observed; the C=C asymmetric stretches with strong intensity that was typical for the alkene group. These results provide polymerization evidence due to the disappearance of double bonds corresponding to monomers and crosslinking agent.
Optical properties
The optical behavior of the hydrogel and its composite was studied by diffuse reflectance (Fig. 3). All these compounds were placed over barium sulfate used as support, due to its low solubility. Figure 3a shows two absorption peaks (blue shift) located approximately at 312–315 and 398–402 nm, respectively, which are attributed to the π–π* and n–π* transitions, corresponding to C=O of the HEMA and DEGDMA. These transitions are also observed in Fig. 3b, c); in these cases, π–π* transition is located with a certain displacement to shorter wavelengths, associated with the presence of silver nanoparticles embedded into hydrogel polymeric matrix [50].

UV/Vis diffuse reflectance spectra for a hydrogel pure; b composite 0.75 M of AgNO3 and c composite 1.0 M of AgNO3
Also, the presence of silver nanoparticles was confirmed via the identification of the typical surface plasmon vibration at a wavelength around 450 nm [51]. The maximum absorption of this band presents a redshift when the AgNO3 concentration was increased from 0.75 to 1 M. The silver nanoparticle surface plasmon changes this position from 425–460 to 445–460 nm, respectively, meaning that the size of nanoparticles increases with the AgNO3 concentration in the solution. This phenomenon could be attributed to the high rate nuclei and therefore with the formation of more silver nanocrystals [52].
Thermal behavior
Differential scanning calorimetry
Thermal analysis shows crystallinity changes in the samples when the silver nanoparticles are added to the solution; also it shows that these are stabilized and embedded into the polymer matrix.
The pure hydrogel with different concentrations of HEMA/DEGDMA has similar glass transition temperature between 110 and 127 °C, values that are concordant with those shown by p-(HEMA) [53]. Thus, when the DEGDMA amount increases (10 %), also does it the Tg to 127 °C, due to physical properties of the crosslinking agent used in the synthesis [54]. This phenomenon is attributed to the crosslinking density imparted by the short-dysfunctional DEGDMA molecules, thus the Tg increase is related principally to the local restriction of coordinated conformational motions at network points. As network density point’s increases, the effective volume of material restricted from coordinated conformational motion also increases, therefore, the material gradually losses the ability to create an appreciable free volume according to temperature changes. This last process produces a glass transition disappearance [55]. In Fig. 4b, composites formed with 0.75 M of AgNO3, show a decrease in the glass transition temperature when HEMA concentration increases and crosslinking agent and DEGDMA concentration decrease. Thus, samples Ag_Hg3-89.5 % until Ag_Hg3-98.5 % present a Tg change from 114 to 103 °C, respectively. This same tendency is exhibited in the Fig. 4c, leaving in evidence that the concentration of silver nitrate does not affect the crystallinity degree of the composites, neither, in considerable way, the glass transition temperature values. These patterns only can be modified by changing the amount of monomer and crosslinking agent in the solution.

Differential scanning calorimetry of a pure hydrogel, b Composite 0.75 M of AgNO3 and c Composite 1.0 M of AgNO3
Thermogravimetric analysis
The hydrogel thermal behavior is considerably affected with the insertion of nanoparticles into the polymer matrix (Fig. 5). The initial decomposition temperature at 5 and 10 % weight losses (T5 % and T10 %) and the charge yields at 500 °C by the alone hydrogel and its composites are summarized in Table 1.

Thermal analysis of a hydrogel pure, b Composite 0.75 M of AgNO3 and c Composite 1.0 M of AgNO3
Figure 5a shows the thermal degradation of the hydrogel alone. These curves show one weight reduction starting at ~250 °C resulting from the degradation of HEMA and eventually the crosslinking agent, DEGDMA. In addition, when the amount of HEMA used by the polymer in their formation increases also does it its thermal stability and residual percentage. This effect is clearly attributed to the generation of free volume into the system. Thus, when a major disorder is produced into the network, caused by a crosslinking agent increase, the compound loses thermal stability. Generally, the increase in thermal stability is related to an increase in the crosslinking agent amount; Farhat et al. demonstrate that with a P(NVP-co-nBMA) compound crosslinked with DEGDMA and TMPTMA, respectively [56]. However, Goiti et al. [57] study the magnetic hydrogel formation based on PVA and PHEMA with magnetite (F3O4), reaching to the conclusion that thermal properties are poorer as the content of crosslinking agent increases. This behavior is similar to the obtained in our samples.
The composites thermal stability (hydrogel–silver nanoparticles) is diminished in comparison to the hydrogel alone (Fig. 5b, c). This decrease could be attributed to the fact that the presence of silver nanoparticles into the hydrogel matrix has the tendency to increase the polymer chains mobility and, therefore, to produce polymer networks with free space increased.
The charge yields are directly attributed to the amount of silver nanoparticles into the system (AgNO3). Thus, when a proportion increases, the inorganic phase of the hydrogel matrix appears with a major residual mass percentage at 500 °C. These studies reveals that embedment of silver nanoparticles reduces thermal weight loss corresponding to polymer degradation step.
XRD patterns
Diffractograms patters were obtained using a CuKα anode with measurements between 5° and 140° with 0.02° steps in the whole interval for two hydrogel nanoparticle samples, both with the same HEMA/DEGDMA ratio (89.5 %) but the first with 0.75 M and the other one with 1.0 M of AgNO3 concentration.
The graph shows the typical wave baseline characteristic to the polymers that do not have a regular crystallographic ordering. The relevant of these results is that the apparition and intensity of crystallographic peaks increase with the AgNO3 concentration. It is important to clarify that these peaks does not correspond to the typical Ag pattern at 2θ = 38.2°, 44.4°, 64.6°, 77.5° and 81.7°, which can be indexed to (111), (200), (220), (311) and (222) planes of pure silver (a = 4.086 Å, JCPDS file No. 04-0783). In this way, is highly probable that these peaks were related with some kind of polymer chain ordering that conforms the hydrogel. These tell us that the presence of silver nanoparticles and the UV exposure produce nucleation and growth of polymer grains around the silver nanoparticles; probably these grains are responsible for the silver nanoparticle stabilization and non-oxidation (Fig. 6).

X-ray diffraction pattern of the composite with different AgNO3 concentrations (0.75 M and 1.0 M)
X-ray fluorescence spectroscopy (XRFS)
This study reveals the chemical composition of the composites (hydrogel nanoparticles). The analysis was performed on pressed pellet; this technique confirmed the presence of silver nanoparticles into the hydrogel matrix. These results are presented in Table 2.
The studies realized with this technique confirm the presence of the elemental crystalline silver. It is important to highlight that this technique only can detect elements with AMU over 22, which means that elements like C, O or H cannot be detected with this technique. These results showed that the percentage of silver in the powder was around 99.5–100 %. However the samples denominated Ag_Hg3-94.5 % (1.0 M), Ag_Hg3-89.5 % (0.75 M) and Ag_Hg3-89.5 % (1.0 M) have traces of sulfur and copper, respectively. These elements could be attributed to raw materials, used in the composite synthesis such as silver nitrate that has traces of sulfate and metallic cooper, among other materials.
X-Ray Photoemission Spectroscopy (XPS)
The XPS data shown in Fig. 7 correspond to the XPS survey of samples, AgHg3-89.5 % (0.75 M) and AgHg3-94.5 % (1 M). These studies show the presence of silver 3d and 3p peaks on the surface due to the formation of Ag nanoparticle on the hydrogel. In addition, carbon and oxygen 1 s peaks correspond to the sample organic structure and also to environmental contamination. In addition, more accurate information could be extracted from high-resolution studies shown in Fig. 8.

Wide scan XPS spectra of Ag_Hg3-89.5 % (0.75 M) and Ag_Hg3-94.5 % (1 M)

XPS high resolution of C 1 s, O 1 s and Ag3d core level for Ag_Hg3-89.5 % (0.75 M) and Ag_Hg3-94.5 % (1 M)
Figure 8 shows C 1 s, O 1 s and Ag 3d spectra of Ag_Hg3-89.5 % (0.75 M) and Ag_Hg3-94.5 % (1 M) composites. For both samples, the C 1 s spectra have been fitted by three peaks corresponding to carbon in different chemical environments. The first peak (blue dashed plot) at 285.0 eV is assigned to –CH3 as terminal group. The peak at 286.2 eV (green dot plot) is possibly associated with alcohol or ether (–C–OH/–C–O–C–), while the third peak (pink solid line) at 288.3 eV can be attributed to ester (–O–C=O) and also to CO2 environmental contamination [58, 59].
The corresponding O 1 s high-resolution spectrum reveals a main peak at 530 eV (pink solid plot) characteristic of metal oxide, in this case attributed to Ag2O encapsulated into the hydrogel and a peak at higher energy (blue dash plot), at 532.2 eV related to metal-OH bonds (–C–O(H)–Ag) [60]. The third component at 533.9 eV (green dot plot) used for fitting the O 1 s spectrum is usually assigned to ester (–C–O–C=O) and the alcohol (–C–OH of the HEMA) [58, 60].
The third column of Fig. 8 corresponds to the Ag 3d core levels, which gives some information about the chemical environment and oxidation state of the silver nanoparticles. As it is possible to observe the 3d splitting from Fig. 8, these peaks are overlapped giving a preliminary idea of mixed silver oxidation states. The curve fitting was performed using 3d silver metallic values (Ag0) letting 3d5/2 at 368.1 eV and 3d3/2 at 374.1 eV; and Ag+1 which let 3d5/2 and 3d3/2 at 368.4 eV and 374.4 eV, respectively [61, 62]. These results predict that the two components of Ag0 and Ag+1 are present in the samples, but due to the small difference between these components, just 0.3 eV [61], it is difficult to distinguish each other. An analysis on the modified Auger parameter (α′) was performed to confirm the mixed chemical states in these samples. The modify Auger parameter is defined as
where Ek is the kinetic energy of the MNN auger peak and EB is the binding energy of 3d5/2 photoelectron peak [63]. The α′ parameter for Ag_Hg3-94.5 % (1 M) was 724.43 eV which is between the values reported for Ag0 and Ag+1, confirming the mixed state predicted by the high-resolution XPS fitting.
From the area of the C 1 s, O 1 s and Ag 3d5/2 it is possible to determine the relative concentration of each element on sample surface; these results are shown in Table 3, where it is possible to observe that the predominant compound is carbon as expected for a polymer and that the concentration of silver is higher for Ag_Hg3-94.5 % (1 M) compared to Ag_Hg3-89.5 % (0.75 M), reaching 3.5 % of relative concentration for the first one.
Morphological analysis (TEM)
Different ratios of HEMA/DEGDMA hydrogels with variations in the AgNO3 concentration were characterized via TEM micrographs. The images were analyzed through counting and diameter measurement of silver nanoparticles in order to obtain a well-populated histogram permitting the visualization of silver nanoparticles diameter distribution, only two of these analysis are showed (Fig. 9). The first one corresponds to an area with several spherical silver nanoparticles with different diameters, but with a clear Gaussian distribution centered at 22.5 ± 7.2 nm, where the associated error corresponds to the standard deviation given by the curve fitting and the diameter distribution. The second image corresponds to an area where a big grain of hydrated hydrogel begins to dry because of the exposures to TEM electron beam. During measurements the samples increase their temperature due to this beam exposure, reaching 40–50 °C in water presence, this effect added to the high vacuum of the TEM chamber produces evaporation of the water occluded between the polymer chains. Therefore, the hydrogel grain began to collapse the releasing silver nanoparticles. In this image (Fig. 9), three different sizes of silver nanoparticles could be identified; the diameter histogram shows three statistical models that correspond to three most repetitive diameters, 37.2 ± 6.1, 85.4 ± 4.9 and 109.8 ± 4.8 nm.

TEM micrograph of the composites Ag_Hg3-89.5 % (0.75 M) and its respective histogram
Figure 10 shows other interesting conformation of the hydrogel/silver nanoparticle system, where it is possible to visualize spherical silver nanoparticles linked between them via the hydrogel matrix, like a bunch. It is important to mention that in this type of conformation the nanoparticle size is very homogeneous instead of the previous case.

TEM micrograph of Ag_Hg3-94.5 % with different AgNO3 concentrations: a 0.75 M and b 1.0 M
Antimicrobial studies
To investigate the biocide action of Ag_Hg3-89.5; Ag_Hg3-94.5; Ag_Hg3-97.0; Ag_Hg3-98.0 and Ag_ Hg3-98.5 % compounds, were performed series of antimicrobial experiments taking E. coli as model bacteria. In early experiments we observed their growth in the presence of Ag_Hg3, by zone inhibition method, taking Hg3 as control sample.
These first approach studies help us to choose Ag_Hg3-89.5 % (1.0 M) as the best compound to perform antimicrobial studies. To realize an accurate measurement of this biocide property, quantitative assays for this sample were performed. Triplicate of the sample Hg3-89.5 % (control) and Ag_Hg3-89.5 % (1.0 M) were prepared according to the procedure. CFU and reduction percentage of E. coli ATCC 25922 against control sample measurements were realized. These results showed a CFU reduction of 99.9 % for this case (Fig. 11). This means that the presence of silver nanoparticles homogeneously distributed into the polymer matrix confers it a very high biocide property against Gram-negative bacteria like E. coli ATCC 25922.

Quantitative assay for E. coli ATCC25922, samples Hg3-89.5 % (control) and Ag_Hg3-89.5 % (1.0 M)
Conclusions
The reduction of Ag+ to Ag0 and the formation of the hydrogel (HEMA/DEGDMA) were performed using a photoinitiator (Irgacure 651). Thus, the novel of the technique lies in the silver nanoparticles in situ formation embedded into hydrogel matrix. This last process confers them spatial and temporal stability due to the high crosslinking degree of the matrix, avoiding oxidation and agglomeration between them. However, as the nanoparticles are surrounded by hydrogel, the pure silver peaks could not be obtained by X-ray diffraction, showing an irregular structure of the polymer. In addition, the UV/Vis spectroscopy detects the surface plasmon vibration characteristic of the silver in a metallic state. Also, TEM was used to visualize nanoparticles with homogenous distribution into the system. XPS proved the existence of silver as Ag° and Ag+ in the composite. Ag Hg3-89.5 % (1.0 M) compound presents an important biocide property reducing 99.9 % of bacterium E. coli ATCC 25922 compared to hydrogel itself (Hg3-89.5 %), used as control.
References
Scognamillo, S., Alzari, V., Nuvoli, D., Mariani, A.: Thermoresponsive super water absorbent hydrogels prepared by frontal polymerization. J. Polym. Sci. A. 48(1), 2486–2490 (2010)
Hoffman, A.S.: Hydrogels for biomedical applications. Adv. Drug Deliver. Rev. 64, 18–23 (2012)
Yu, L., Zhang, Z., Zhang, H., Ding, J.: Biodegradability and biocompatibility of thermoreversible hydrogels formed from mixing a sol and a precipitate of block copolymer in water. Biomacromolecules 11(8), 2169–2178 (2010)
Qiu, Y., Park, K.: Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliver. Rev. 64, 49–60 (2012)
Wichterle, O., Lim, D.: Hydrophilic gels for biological use. Nature 185, 117–118 (1960)
Hoffman, A.S., Schmer, G., Harris, C., Kraft, W.G.: Covalent binding of biomolecules to radiation-grafted hydrogels on inert polymer surfaces. T. Am. Soc. Art. Int. Org. 18, 10–17 (1972)
Ratner, B.D., Hoffman, A.S.: Synthetic hydrogels for biomedical applications, in hydrogels for medical and related applications. ACS Sym. Ser. 31, 1–36 (1976)
Peppas, N.A.: In Hydrogels in Medicine and Pharmacy, vol. 1, pp. 95–126. CRC Press, Boca Raton (1987)
Park, K., Shalaby, W.S.W., Park, H.: Biodegradable Hydrogels for Drug Delivery. Technomic, Lancaster, PA, ch. 9, pp. 233–238 (1993)
Harland, R.S., Prud’homme, R.K.: Polyelectrolyte Gels: Properties, Preparation and Applications, American Chemical Society, Washington, DC, vol. 480, ch. 16, pp. 269–284 (1992)
Ulbrich, K., Subr, V., Podperová, P., Buresová, M.: Synthesis of novel hydrolytically degradable hydrogels for controlled drug. J. Control. Release. 34, 155–165 (1995)
Hoffman, A.S.: Intelligent polymers in medicine and biotechnology. Macromol. Symp. 98, 645–664 (1995)
Harris, J.M., Zalipsky, S.: Poly(ethylene glycol) Chemistry and Biological Applications. ACS Sym. Ser, Washington DC (1997)
Su, J., Hu, B.H., Lowe, W.L., Kaufman, D.B., Messersmith, P.B.: Anti-inflammatory peptide-functionalized hydrogels for insulin-secreting cell encapsulation. Biomaterials 31(2), 308–314 (2010)
Kumachev, A., Greener, J., Tumarkin, E., Eiser, E., Zandstra, P.W., Kumacheva, E.: High-throughput generation of hydrogel microbeads with varying elasticity for cell encapsulation. Biomaterials 32(6), 1477–1483 (2011)
Phelps, E.A., Enemchukwu, N.O., Fiore, V.F., Sy, J.C., Murthy, N., Sulchek, T.A., Barker, T.H., Garcia, A.J.: Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv. Mater. 24(1), 64–70 (2012)
Liu, K., Deng, Y., Zhang, N., Li, S., Ding, H., Guo, F., Liu, W., Guo, S., Zhao, X.Z.: Generation of disk-like hydrogel beads for cell encapsulation and manipulation using a droplet-based microfluidic device. Microfluid. Nanofluid. 13(5), 761–767 (2012)
Zhu, J.: Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 31(17), 4639–4656 (2010)
Van Vlierberghe, S., Dubruel, P., Schacht, E.: Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review. Biomacromolecules 12(5), 1387–1408 (2011)
Tan, H., Chu, C.R., Payne, K.A., Marra, J.G.: Injectable in situ forming biodegradable chitosan-hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials 30(13), 2499–2506 (2009)
Zhu, J., Marchant, R.E.: Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devic. 8(5), 607–626 (2011)
Kostina, N.Y., Sharifi, S., De Los Santos Pereira, A., Michálek, J., Grijpma, D.W., Rodriguez-Emmernegger, C.: Novel antifouling self-healing poly(carboxybetaine methacrylamide-co-HEMA) nanocomposite hydrogels with superior mechanical properties. J. Mater. Chem. 41, 5644–5650 (2013)
Gawel, K., Gao, M., Stokke, B.T.: Impregnation of weakly charged anionic microhydrogels with cationic polyelectrolytes and their swelling properties monitored by a high resolution interferometric technique. Transformation from a polyelectrolyte to polyampholyte hydrogel. Eur. Polym. J. 48(11), 1949–1959 (2012)
Vilozny, B., Schiller, A., Wessling, R.A., Singaram, B.: Multiwell plates loaded with fluorescent hydrogel sensors for measuring pH and glucose concentration. J. Mater. Chem. 21, 7589–7595 (2011)
Kali, G., Vavra, S., László, K., Iván, B.I.: Thermally responsive amphiphilic conetworks and gels based on poly(N-isopropylacrylamide) and polyisobutylene. Macromolecules 46(13), 5337–5344 (2013)
Huang, H., Li, W., Wang, H., Zeng, X., Wang, Q., Yang, Y.: Conducting hydrogels of tetraaniline-g-poly(vinyl alcohol) in situ reinforced by supramolecular nanofibers. ACS Appl. Mater. Interfaces. 6(3), 1595–1600 (2014)
Pafiti, K.S., Elladiou, M., Patrickios, C.S.: “Inverse polyampholyte” hydrogels from double-cationic hydrogels: synthesis by RAFT polymerization and characterization. Macromolecules 47(5), 1819–1827 (2014)
Way, A.E., Korpusik, A.B., Dorsey, T.B., Buerkle, L.E., Von Recum, H.A., Rowan, S.J.: Enhancing the mechanical properties of guanosine-based supramolecular hydrogels with guanosine-containing polymers. Macromolecules 47(5), 1810–1818 (2014)
Qingzhou, C., Wei, W., Baohua, G., Liyuan, L.: A Combined physical-chemical polymerization process for fabrication of nanoparticle-hydrogel sensing materials. Macromolecules 45(20), 8382–8386 (2012)
Tuncaboylu, D.C., Sahin, M., Argun, A., Oppermann, W., Okay, O.: Dynamics and large strain behavior of self-healing hydrogels with and without Surfactants. Macromolecules 45(4), 1991–2000 (2012)
Yue, Y.M., Sheng, X., Wang, P.X.: Fabrication and characterization of microstructured and pH sensitive interpenetrating networks hydrogel films and application in drug delivery field. Eur. Polym. J. 45(2), 309–315 (2009)
Guiseppi-Eliea, A.: Electroconductive hydrogels: synthesis, characterization and biomedical applications. Biomaterials 31(10), 2701–2716 (2010)
Jagur-Grodzinski, J.: Polymeric gels and hydrogels for biomedical and pharmaceutical applications. Polym. Adv. Technol. 21(1), 27–47 (2010)
Baït, N., Grassl, B., Derail, C., Benaboura, A.: Hydrogel nanocomposites as pressure-sensitive adhesives for skin-contact applications. Soft Matter 7, 2025–2032 (2011)
Li, L., Lee, L.J.: Photopolymerization of HEMA/DEGDMA hydrogels in solution. Polymer 46(25), 11540–11547 (2005)
Otsuka, H., Nagasaki, Y., Kataoka, K.: PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 64, 246–255 (2012)
Petros, R.A., DeSimone, J.M.: Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 9(8), 615–627 (2010)
Sau, T.K., Rogach, A.L., Jäckel, F., Klar, T.A., Feldmann, J.: Properties and applications of colloidal nonspherical noble metal nanoparticles. Adv. Mater. 22(16), 1805–1825 (2010)
Hao, R., Xing, R., Xu, Z., Hou, Y., Gao, S., Sun, S.: Synthesis, functionalization and biomedical applications of multifunctional magnetic nanoparticles. Adv. Mater. 22(25), 2729–2742 (2010)
Jain, J., Arora, S., Rajwade, J.M., Omray, P., Khandelwal, S., Paknikar, K.M.: Silver nanoparticles in therapeutics: development of an antimicrobial gel formulation for topical use. Mol. Pharm. 6(5), 1388–1401 (2009)
Verma, V.C., Kharwar, R.N., Gange, A.C.: Biosynthesis of antimicrobial silver nanoparticles by the endophytic fungus Aspergillus clavatus. Nanomed. UK 5(1), 33–40 (2010)
Singhal, G., Bhavesh, R., Kasariya, K., Sharma, A.R., Singh, R.P.: Biosynthesis of silver nanoparticles using Ocimum sanctum (Tulsi) leaf extract and screening its antimicrobial activity. J. Nanopart. Res. 13(7), 2981–2988 (2011)
Rai, M., Yadav, A., Gade, A.: Silver nanoparticles as a new generation of antimicrobials. Biotechnol. Adv. 27(1), 76–83 (2009)
Sharma, V.K., Yngard, R.A., Lin, Y.: Silver nanoparticles: green synthesis and their antimicrobial activities. Adv. Colloid. Interface. 145(1–2), 83–96 (2009)
Kim, S.H., Lee, H.S., Ryu, D.S., Choi, S.J., Lee, D.S.: Antibacterial activity of silver-nanoparticles against Staphylococcus aureus and Escherichia coli. Korean J. Microbiol. Biotechnol. 39(1), 77–85 (2011)
Varsha, T., Bajpai, M., Bajpai, S.K.: In situ formation of silver nanoparticles within Chitosan-attached cotton fabric for antibacterial property. J. Ind. Text. 40(3), 229–245 (2011)
Agnihotri, S., Mukherji, S., Mukherji, S.: Antimicrobial chitosan–PVA hydrogel as a nanoreactor and immobilizing matrix for silver nanoparticles. Appl Nanosci. 2(3), 179–188 (2012)
Vermonden, T., Censi, R., Hennink, W.E.: Hydrogels for protein delivery. Chem. Rev. 112(5), 2853–2888 (2012)
Lin-Vien, D., Colthup, N.B., Fateley, W.G., Grasselli, J.G.: The handbook of infrared and Raman characteristic frequencies of organic molecules. Academic Press, San Diego (1991). 74
Cook, W.D., Nghiem, Q.D., Chen, Q., Chen, F., Sangermano, M.: Simultaneous photoinduced silver nanoparticles formation and cationic polymerization of divinyl ethers. Macromolecules 44(11), 4065–4071 (2011)
Guzman, M., Dille, J., Godet, S.: Synthesis and antibacterial activity of silver nanoparticles against gram-positive and gram-negative bacteria. Nanomed-Nanotechnol. 8, 37–45 (2012)
Fendler, J.H., Dékány, I.: Nanoparticles in solid and solutions. Springer, New York (1997). 117
Yao, F., Xu, L.Q., Fu, G.D., Lin, B.P.: Sliding-graft interpenetrating polymer networks from simultaneous “Click Chemistry” and atom transfer radical polymerization. Macromolecules 43(23), 9761–9770 (2010)
Uygun, M., Cook, W.D., Moorhoff, C., Chen, F., Vallo, C., Yagci, Y., Sangermano, M.: Photopolymerization kinetics and dynamic mechanical properties of silanes hydrolyzed without evolution of byproducts. Tetrakis[(methacryloyloxy)ethoxy]silane−Diethylene glycol dimethacrylate. Macromolecules 44(7), 1792–1800 (2011)
Wornyo, E: Fabrication and Characterization of Shape Memory Polymers at Small-scales, ProQuest, p. 51. UMI Dissertation Publishing, Ann Harbor, Michigan, USA (2011)
Huma, F., Akhter, Z., Yasin, T., Zafar-uz-Zaman, M., Manan, A.: Crosslinking of poly(N-vinyl pyrrolidone-co-n-butyl methacrylate) copolymers for controlled drug delivery. Polym. Bull. 71, 433–445 (2014)
Goiti, E., Salinas, M.M., Arias, G., Puglia, D., Kenny, J.M., Mijangos, C.: Effect of magnetic nanoparticles on the thermal properties of some hydrogels. Polym. Degrad. Stabil. 92, 2198–2205 (2007)
Carrol, K.J., Reveless, J.U., Shultz, M.D., Khanna, S.N., Carpenter, E.E.: Preparation of elemental Cu and Ni nanoparticles by the polyol method: an experimental and theoretical approach. J. Phys. Chem. C 115(6), 2656–2664 (2011)
Prieto, P., Nistor, V., Nouneh, K., Oyama, M., Abd-Lefdil, M., Díaz, R.: XPS study of silver, nickel and bimetallic silver-nickel nanoparticles prepared by seed-mediated growth. Appl. Surf. Sci. 258(22), 8807–8813 (2012)
Jeon, M.K., Kang, M.: Synthesis and characterization of indium-tin-oxide particles prepared using sol–gel and solvothermal methods and their conductivities after fixation on polyethyleneterephthalate films. Mater. Lett. 62(4–5), 676–682 (2008)
Ferraria, A.M., Carapeto, A.P., Botelho do Rego, A.M.: X-ray photoelectron spectroscopy: silver salts revisited. Vacuum 86(12), 1988–1991 (2012)
Sumesh, E., Bootharaju, M.S., Pradeep, A.T.: A practical silver nanoparticle-based adsorbent for the removal of Hg2+ from water. J. Hazard. Mater. 189(1–2), 450–457 (2011)
Satta, M., Moretti, G.: Auger parameters and Wagner plots. J Electron Spectrosc. 178, 123–127 (2010)
Acknowledgments
The authors acknowledge the financial support of this work by FONDECYT Grant No 11121281 and 1110836. Dr. Gonzalez acknowledge to the attraction and insertion of advanced human capital program, PAI 7912010031, CONICYT. Mr. Sarabia acknowledges the financial support given by CONYCYT through the Magister Scholarship Grant. Ms. Susana Rojas acknowledges the financial support given by CONYCYT through the Doctoral Scholarship Grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Henríquez, C.M.G., del Carmen Pizarro Guerra, G., Vallejos, M.A.S. et al. In situ silver nanoparticle formation embedded into a photopolymerized hydrogel with biocide properties. J Nanostruct Chem 4, 119–132 (2014). https://doi.org/10.1007/s40097-014-0125-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-014-0125-y