
Abstract
In this study, we investigate the adsorption properties of hydrogen florid (HF) and sulfur trioxide (SO3) on the surface of platinum decorated graphene (PtG) using density functional theory. We found one optimized configuration for HF and two ones for SO3 upon adsorption on the surface of PtG. Our result show significant adsorption on PtG with calculated energy adsorption of −73.6 (−54.2 BSSE) kJ/mol for HF at its only position and −172.4 (−144.8 BSSE) and −62.7 (−53.7 BSSE) kJ/mol for SO3 at its two positions; P1 and P2, respectively), whereas there is weak physisorption of these analytes on pristine graphene (PG). Results of charge analyses reveled interesting net charge transfer; while the direction of charge is from HF to PtG, reverse direction is found for SO3 for its two configurations. To deep understand the concept of adsorption properties, we used orbital analyses including density of states for interaction of mentioned analytes on the surface of PtG.
Similar content being viewed by others

Introduction
Graphene has connected carbons in the array of two-dimensional sp2. It has excellent properties such as high surface-to-volume fraction [1]. Due to its marvelous electronic structure, it has been used for numerous times as gas sensor for different analytes [2, 3]. All graphene-made electronic devices benefit from its exceptional charge transferring and its high steadiness. Along with carbon nanotubes [4, 5], there are many experimental and theoretical studies focused on graphene for sensing materials [6–8]. Some research works on graphene as gas sensor were listed and discussed by Mao et al. [9]. They disputed on the opportunities of utilizing these materials for gas sensor purpose.
The properties of graphene could be improved particularly by modification of its surface by doping and decoration of different hetro atoms [10–13]. In our recent study, we have reported the potential of N-doped graphene towards its interaction with boron [14], and SOx [15] and CO [16]. We documented that this modified surface is able to increase the adsorption properties of these compounds. Moreover, our group reported different application of Al and B-doped graphene sheets [17–24]. It has been proved in these papers that metal doping by Al or B increases the adsorbent potential of graphene for practical application.
Besides doping process, metal decoration is another way to intensify the adsorption property of graphene. Upon decoration, single atom could be placed on high surface area carbons. It will induce to addition of several re-bonding per metal atom with releasing significant energy, which consequents more steadiness of single atom decorated structure in comparison with bulk atoms decorated structure [25]. There are diverse experimentally techniques for single atom decoration on nanostructures. As an example, Wang et al. [26] showed that a two-step process is a well-ordered technique to stabilize single atoms on graphene. Diverse metals including Pt, Co, and In, have been successfully incorporated in graphene in the single-atom form. As an example, Baby et al. [27] built an amperometric glucose biosensor by decoration of Pt and Au nano-particles on graphene. Based on their research, decorated particles could able to immobilize glucose oxidase by physical adsorption follow-on an enhancement in the performance of biosensor. Moreover, our group searched on the adsorption of NO molecule on the surface of Pt-decorated graphene [28]. We found very high adsorption on this modified surface while there was weak adsorption in the case of using pristine graphene. In parallel we used Pt-decorated graphene as an ideal adsorbent for C2H2 and C2H4 molecules [29], MeOH and EtOH [30], and SO2 and O3 [31].
Hydrogen fluoride (HF) is a colorless gas and main industrial source of fluorine, frequently as an aqueous solution named hydrofluoric acid. It is the forerunner for different significant compounds such as polymers (for example Teflon). HF is extensively utilized in the petrochemical industry as a part of super acids. The adsorption of HF on the surface of pristine graphene has been deeply investigated by Sun et al. [32]. They used various configurations for adsorption of HF using DFT and found that at its most energetically favorable site, the amount of adsorption energy is very weak (physisorption).
On the other hand, sulfur trioxide (SO3) in the gaseous form is a vital pollutant, which is the primary agent in acid rain. It has been produced on an industrial scale as a precursor to sulfuric acid. Adsorption of SO3 on pristine graphene has been investigated previously by our group [15]. We found there is very weak adsorption of SO3 on pristine graphene.
In this study, we select PtG sheet to have more investigation since its potential as adsorbent is well proven. Here we aim to establish the potential of PtG as a new adsorbent for HF and SO3 molecules by using first-principles simulation. The gas molecules HF and SO3 are of pollution for industrial as well as environmental applications. They are very poison gases and so there is much require to their removal at industrial situations. To the best of our knowledge, there is no reported study on the adsorption properties of HF and SO3 molecules on the surface of PtG. The inspiration of this study is to attain basic insights to the impact of adsorbed HF and SO3 molecules on the electronic structure of PtG.
Computational method
All relaxation for PG and PtG sheets in the present and absence of HF and SO3 molecules were done using B3LYP density functional with split basis set (for all atom excluding Pt the basis set was 6-31G (d,p), for Pt atom the basis set was lanl2dz) as implemented in Gaussian 09 suite of program [33]. To save the time, initial optimizations were carried by using 3-21G(d)/lanl2dz basis set (for all possible configurations) to distinguish the most stable configurations. Then, among all relaxed structures achieved by this initial functional/basis set, those having highest stability (judging from their calculated adsorption energy) were selected as input files to do next optimization at stronger basis set/functional (6-31G(d,p)/B3LYP) to achieve more precise values of parameters (adsorption energy, equilibrium distance, charge transfer,…).
The 6-31G(d,p) basis set is good for general calculations, besides the B3LYP density functional has been known appropriate for nano-structure studies [28–31].
All Calculations including charge analysis, density of states (DOS), the energy of lowest unoccupied molecular orbital (LUMO), the energy of highest occupied molecular orbital (HOMO), and the HOMO–LUMO energy gap (E g) have been done using above-mentioned level of theory.
The values of adsorption energy (E ads) upon adsorption of analyte on PtG were specified using Eq. (1):
where the E PtG-A corresponds to the adsorbed system of PtG, E PtG correspond to the isolated PtG and E A corresponds to the isolated analyte (HF or SO3), respectively. For all adsorbed systems, calculated adsorption energies were modified based on the Eq. (2).
which E ads, CP is counterpoise corrected adsorption energy of related complexes and E BSSE is basis set superposition errors energy.
Result and discussions
A supercell of 4 × 4 graphene (12.30 × 12.30 × 16 Å) (left side of Fig. 1) has been used as surface of interaction. There is no interaction between graphene sheets of neighboring supercells since the z axis (16 Å) of this supercell is big enough.

A supercell of graphene (left) and three possible adsorption sites for decoration of Pt (right)
As can be seen in the right side of Fig. 1, there is three potential sites for Pt decoration on graphene: the bridge site (considering the middle of a C–C bond), the top site (directly above a C atom) and the hollow site (at the middle of a hexagon). As we reported in our recent publications [28–31], the bridge site of graphene is the most energetically position for the decoration of Pt compared to the two other ones. Therefore, we placed the Pt atom on this site and let this initial configuration to be optimized at the above-mentioned basis set/functional to made PtG.
As mentioned in the section of introduction, the adsorption properties of both HF and SO3 on the surface of pristine graphene were investigated before [15, 32] and their relaxed structure were well discussed. For both analytes, pristine graphene revealed very low adsorption (−7.1 kJ/mol for HF and −9.1 kJ/mol for SO3, respectively). For this reason, we disregarded to repeat the study of adsorption of these analytes on pristine graphene.
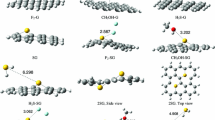
To find the most stable configurations of HF and SO3 on Pt-decorated graphene, first we made all possible configurations as input files. For HF, it was placed on top of Pt atom so that the H-F axis in one state has parallel configuration and in other states has perpendicular configurations (one, from the H-side and another, from the F-side) than the surface. Moreover, we placed SO3 on top of Pt from the O-side (perpendicular to the surface) and the S-side (parallel to the surface). All these initial configurations were subjected to relaxation at minimum basis set of 3-21G(d) (lanl2dz for Pt) using DFT to primary finding of relaxed structures. We found that all three input structures for HF turn to only one position upon optimization, whereas there are two relaxed structures for output of SO3-including systems. Additionally, we used these initial optimized structures (one for HF and two for SO3) as input files for next optimization at the stronger basis set of 6-31G (d,p)/(B3LYP, lanl2dz for Pt) to get more precise structure properties. Figure 2 represents the relaxed structures of these adsorbed systems.

Side views (left) and top views (right) of some parts of relaxed structure of adsorbed HF (a) and SO3 [P1 (b) and P2 (c)] on PtG
Relaxation of HF on PtG accomplishes with releasing −73.6 kJ/mol with closest equilibrium distance of 2.27 Å (see Fig. 2). These values are considerable compared to those reported for HF adsorption on pristine graphene (the adsorption energy of −7.1 kJ/mol and adsorption distance of 3.66 Å) [32]. Moreover, fully optimization of SO3 on PtG corresponds to releasing energies of −172.4 and −62.7 kJ/mol for position 1 and 2, respectively (P1 and P2). The adsorption value of SO3 on PtG in both configuration are much higher compared to that reported on pristine graphene (−9.1 kJ/mol) [15]. The closest adsorption distances are achieved 2.16 and 2.00 Å for P1 and P2, respectively, which are completely shorter compared to its adsorption on pristine graphene (3.25 Å) [15]. Despite P1 represents much higher adsorption energy compared to P2, however, the closest adsorption distance of the later is relatively lower than that of P1. This is because in P1, all atoms of SO3 involve in formation of bond with Pt, while in P2, only one atom (O) interacts with Pt.
We can conclude that interactions of HF and SO3 (in P1 and P2) on PtG could be categorized in chemisorption region. These extensive higher values of HF and SO3 adsorption on PtG than their adsorption on PG is a result of high interaction between Pt…F and Pt…O upon their interaction on the earlier surface.
Then, we investigate on the net charge transfer for adsorption of HF and SO3 (P1 and P2) on the surface of PtG using natural bond orbital (NBO). The data are listed in Table 1. The net charge transfer for adsorption of HF and SO3 were calculated +0.171, −0.337 (P1), and −0.364 (P2) e, respectively. Reverse direction of charge transfer for HF compared to SO3 points to the ability of PtG to act as n-type and p-type semiconductors at the same time. This can be confirmed by considering the local charge of Pt upon adsorption of these analytes (−0.101 e for HF, +0.236 e for SO3 (P1), and +0.258 e for SO3 (P2), as can be seen in Table 1).
These results accentuate that the adsorption of HF and SO3 on PtG significantly change its electronic property.
To examine the change in the electronic structure of graphene resulted by decoration of Pt, the HOMO and LUMO distributions of pristine and Pt-decorated graphene sheets are depicted in Fig. 3. It can be seen in Fig. 3, that the HOMO of PG is mostly restricted on the C–C bonds whereas the LUMO is situated on the conflicting site. After Pt is decorated on graphene, major parts of both HOMO and LUMO relocate on Pt atom which result more reactivity of PtG compared to PG. Additionally, the result of NBO analysis showed that the C-atoms neighboring Pt attract charges owing to their high electron affinity and then result a reduce (+0.132 e) in the electron density of Pt.

HOMO and LUMO distributions of different systems
The HOMO and LUMO of adsorbed system for complexed PtG are portrayed in Fig. 3 and the data are listed in Table 1. By comparing the HOMO–LUMO distributions of free PtG with those of adsorbed ones, it can be found that the distribution of both HOMO and LUMO are effected by adsorbate. These effects are more pronounced in the case of SO3 rather than HF as can be expected by their difference in the calculated values of adsorption energy. By considering the data of Table 1, we can say that upon adsorption of HF, increase in the energies of HOMO and LUMO of system from −5.00 and −2.58 eV to −4.92 and −1.84 eV will happen, respectively. This increase associates with transfer of charge from HF to PtG (p-type semiconductor). On the other hand, for SO3 adsorption, the energies of HOMO and LUMO of system decrease to −5.85 and −2.89 eV for P1 and −5.49 and −4.04 eV from their initial values (listed in Table 1). These changes accomplishes with transfer of charge from PtG to SO3 (n-type semiconductor).
To deep understanding of the electronic property of PtG upon adsorption of HF and SO3, the DOSs for all systems in free (Fig. 4a, b) and complexed forms were depicted close to the Fermi level (see Fig. 4). Comparing the DOSs of free PtG to that of PtG-HF complex (Fig. 4c, d) we can find significant changes in the location of HOMO and LUMO, in which the E g (E HOMO − E LUMO) of system increase from 2.42 to 3.08 eV. This increase in the E g corresponds to increase in the stability of system upon adsorption [34–36].

DOSs for free HF (a), free SO3 (b), free PtG (c), PtG-HF complex (d), PtG-SO3 (position 1) (e), and PtG-SO3 (position 2) (f)
In the other hand, by comparing the DOSs of free PtG (Fig. 4c) with PtG-SO3 (Fig. 4c, e, f) important change confirmation of hybridization between SO3 and PtG (in both positions) could be find upon adsorption which introduce this modified surface as excellent adsorbent for SO3.
Conclusion
In this study, we search the adsorption of HF and SO3 molecules on the surfaces of PtG. First, we calculated the energy of any relaxed system followed by calculation of orbital descriptions, DOSs and NBO analyses. It was found that the interaction of HF and SO3 molecules on PtG changes its electronic structure. The reason for our investigation on PtG was based on knowing this matter of fact that pristine graphene has very weak interaction with these molecules. Decoration of graphene by Pt considerably enhances the potential of graphene to be interacted. The NBO charge analysis reveals reverse direction of charge transfer for SO3 compared to HF, pointing PtG is n-type and p-type semiconductors at the same time.
References
Gadipelli, S., Xiao, G.Z.: Graphene-based materials: synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 69, 1–60 (2015)
Schedin, F., Geim, A.K., Morozov, S.V., Hill, E.W., Blake, P., Katsnelson, M.I., Novoselov, K.S.: Detection of individual gas molecules adsorbed on graphene. Nat. Mater. 6, 652–655 (2007)
Barbolina, I.I., Novoselov, K.S., Morozov, S.V., Dubonos, S.V., Missous, M., Volkov, A.O., Christian, D.A., Grigorievam, I.V., Geim, A.K.: Submicron sensors of local electric field with single-electron resolution at room temperature. Appl. Phys. Lett. 88, 013901 (2006)
Majzlíkova, P., Sedlacek, J., Prasek, J., Pekarek, J., Svatos, V., Bannov, A.G., Jasek, O., Synek, P., Elias, M., Zajickova, L., Hubalek, J.: Sensing properties of multiwalled carbon nanotubes grown in MW plasma torch: electronic and electrochemical behavior, gas sensing, field emission, ir absorption. Sensors 15, 2644–2661 (2015)
Liu, S.F., Petty, A.R., Sazama, G.T., Swager, T.M.: Single-walled carbon nanotube/metalloporphyrin composites for the chemiresistive detection of amines and meat spoilage. Angew. Chem. Int. Ed. 54, 6554–6557 (2015)
Leenaerts, O., Partoens, B., Peeters, F.M.: Adsorption of H2O, NH3, CO, NO2, and NO on graphene: a first-principles study. Phys. Rev. B 77, 125416 (2008)
Ao, Z.M., Yang, J., Li, S., Jiang, Q.: Enhancement of CO detection in Al doped graphene. Chem. Phys. Lett. 461, 276–279 (2008)
Kang, H.S.: Theoretical study of binding of metal-doped graphene sheet and carbon nanotubes with dioxin. J. Am. Chem. Soc. 127, 9839–9843 (2005)
Mao, S., Lu, G., Chen, J.: Nanocarbon-based gas sensors: progress and challenges. J. Mater. Chem. A 2, 5573–5579 (2014)
Zacharia, R., Kim, K.Y., Kibria, A.K.M.F., Nahm, K.S.: Enhancement of hydrogen storage capacity of carbon nanotubes via spill-over from vanadium and palladium nanoparticles. Chem. Phys. Lett. 412, 369–375 (2005)
Kim, H.S., Lee, H., Han, K.S., Kim, J.H., Song, M.S., Park, M.S., Lee, J.Y., Kang, J.K.: Hydrogen storage in Ni nanoparticle-dispersed multiwalled carbon nanotubes. J. Phys. Chem. B 109, 8983–8986 (2005)
Li, Y.W., Yang, R.T.: Significantly enhanced hydrogen storage in metal-organic frameworks via spillover. J. Am. Chem. Soc. 128, 726–727 (2006)
Gao, S., Ren, Z., Wan, L., Zheng, J., Guo, P., Zhou, Y.: Density functional theory prediction for diffusion of lithium on boron-doped graphene surface. Appl. Surf. Sci. 257, 7443–7446 (2011)
ShokuhiRad, A., Shadravan, A., Soleymani, A.A., Motaghedi, N.: Lewis acid-base surface interaction of some boron compounds with N-doped graphene; first principles study. Curr. Appl. Phys. 15, 1271–1277 (2015)
ShokuhiRad, A., Esfahanian, M., Maleki, S., Gharati, G.: Application of carbon nanostructures towards SO2 and SO3 adsorption: a comparison between pristine graphene and N-doped graphene by DFT calculations. J. Sulfur Chem. 37, 176–188 (2016)
ShokuhiRad, A., Shabestari, S.S., Jafari, S.A., Zardoost, M.R., Mirabi, A.: N-doped graphene as a nanostructure adsorbent for carbon monoxide: DFT calculations. Mol. Phys. 114, 1756–1762 (2016)
ShokuhiRad, A., Kashani, O.R.: Adsorption of acetyl halide molecules on the surface of pristine and Al-doped graphene: ab initio study. Appl. Surf. Sci. 355, 233–241 (2015)
ShokuhiRad, A.: Al-doped graphene as sensitive nanostructure sensor for some ether molecules: ab initio study of adsorption. Synth. Met. 209, 419–425 (2015)
ShokuhiRad, A.: First principles study of Al-doped graphene as nanostructure adsorbent for NO2 and N2O: DFT calculations. Appl. Surf. Sci. 357, 1217–1224 (2015)
ShokuhiRad, A., Foukolaei, V.P.: Density functional study of Al-doped graphene nanostructure towards adsorption of CO, CO2 and H2O. Synth. Met. 210, 171–178 (2015)
ShokuhiRad, A.: Al-doped graphene as a new nanostructure adsorbent for some halomethane compounds: DFT calculations. Surf. Sci. 645, 6–12 (2016)
ShokuhiRad, A., Jouibary, Y.M., Foukolaei, V.P., Binaeian, E.: Study on the structure and electronic property of adsorbed guanine on aluminum doped graphene: first principles calculations. Curr. Appl. Phys. 16, 527–533 (2016)
ShokuhiRad, A., Shabestari, S.S., Mohseni, S., Aghouzi, S.A.: Study on the adsorption properties of O3, SO2, and SO3 on B-doped graphene using DFT calculations. J. Solid State Chem. 237, 204–210 (2016)
ShokuhiRad, A.: Adsorption of mercaptopyridine on the surface of Al- and B-doped graphenes: theoretical study. J. Alloy. Compd. 682, 345–351 (2016)
Contescu, C.I., van Benthem, K., Li, S., Bonifacio, C.S., Pennycook, S.J., Jena, P., Gallego, N.C.: Single Pd atoms in activated carbon fibers and their contribution to hydrogen storage. Carbone 49, 4050–4058 (2011)
Wang, H., Wang, Q., Cheng, Y., Li, K., Yao, Y., Zhang, Q., Dong, C., Wang, P., Schwingenschlögl, U., Yang, W., Zhang, X.X.: Doping monolayer graphene with single atom substitutions. Nano Lett. 12, 141–144 (2012)
Baby, T.T., Aravind, S.S.J., Arockiadoss, T., Rakhi, R.B., Ramaprabhu, S.: Metal decorated graphene nanosheets as immobilization matrix for amperometric glucose biosensor. Sensor Actuat. B Chem. 145, 71–77 (2010)
ShokuhiRad, A., Abedini, E.: Chemisorption of NO on Pt-decorated graphene as modified nanostructure media: a first principles study. Appl. Surf. Sci. 360, 1041–1046 (2016)
ShokuhiRad, A.: Adsorption of C2H2 and C2H4 on Pt-decorated graphene nanostructure: ab initio study. Synth. Met. 211, 115–120 (2016)
ShokuhiRad, A.: Density functional theory study of the adsorption of MeOH and EtOH on the surface of Pt-decorated graphene. Physica E 83, 135–140 (2016)
ShokuhiRad, A., Zareyee, D.: Adsorption properties of SO2 and O3 molecules on Pt-decorated graphene: a theoretical study. Vacuum 130, 113–118 (2016)
Sun, Y., Chen, L., Zhang, F., Li, D., Pan, H., Ye, J.: First-principles studies of HF molecule adsorption on intrinsic graphene and Al-doped graphene. Solid State Commun. 150, 1906–1910 (2010)
Frisch, M.J., et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT (2009)
ShokuhiRad, A., Ayub, K.: A comparative density functional theory study of guanine chemisorption on Al12N12, Al12P12, B12N12, and B12P12 nano-cages. J. Alloy. Compd. 672, 161–169 (2016)
ShokuhiRad, A., Ayub, K.: Detailed surface study of adsorbed nickel on Al12N12 nano-cage. Thin Solid Films 612, 179–185 (2016)
ShokuhiRad, A., Ayub, K.: Ni adsorption on Al12P12 nano-cage: DFT study. J. Alloy. Compd. 678, 317–324 (2016)
Acknowledgments
I highly acknowledge financial support from Iran Nanotechnology Initiative Council, Iran.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rad, A.S. DFT study of hydrogen fluoride and sulfur trioxide interactions on the surface of Pt-decorated graphene. J Theor Appl Phys 10, 307–313 (2016). https://doi.org/10.1007/s40094-016-0230-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40094-016-0230-z