
Abstract
Homo- and copolymers of 2-(N-phthalimido)ethyl methacrylate (NPEMA) and p-chlorophenyl methacrylate (PCPMA) were prepared in N,N-dimethyl formamide (DMF) solution at 70 °C using 2,2-azo-bisisobutyronitrile (AIBN) as initiator. The nano-CdS-doped polymer composite of NPEMA and PCPMA was prepared via in situ technique. The homo- and copolymers of NPEMA and PCPMA were characterized using FT-IR spectroscopy and gel permeation chromatography (GPC). The polymer nano composites were characterized using FT-IR spectroscopy, X-ray diffraction, and transmission electron microscopy. The reactivity ratios (r1 and r2) were obtained from the various linear graphical methods. The values of r1 (NPEMA) = 0.55 and r2 (PCPMA) = 1.30 were found from the same graphical methods. The copolymer microstructures were found from the mean sequence length, run number, and dyad fraction. Thermal behavior of polymers and polymer nano composites under nitrogen atmosphere was studied. The activation energies of neat polymers were varied in the range of 56–85 kJ/mol, while 28–56 kJ/mol energies were found for nano-CdS-doped polymer composites. The thermodynamic parameters of thermal degradation were also obtained. Kinetic and thermodynamic parameters were confirming the stability of the neat polymers than polymer nano composites. The polymers were assessed on different microorganisms for obtaining the antimicrobial properties. Overall, the polymers permit 10–52, 20–58, and 18–56% growth of bacteria, fungi, and yeast, respectively.
Similar content being viewed by others

Introduction
Copolymerization technique is an adaptable technique to prepare copolymers from two or more different types of monomers. These copolymers have different properties than their individual homopolymers. The acrylates or methacrylates are normally polymerized through various techniques. These techniques are free radical solution polymerization [1], bulk polymerization [2], emulsion polymerization [3], atom transfer free radical polymerization (ATRP) [4], reversible addition–fragmentation chain transfer free radical polymerization (RAFT) [5], redox polymerization [6], photo polymerization [7] living polymerization [8], etc. It is required to choose the polymerization process to obtain copolymers having desirable chemical, optical, mechanical, and electrical properties. Free radical solution polymerization is a simple, versatile, and effective technique to prepare the acrylate or methacrylate homo- and copolymers. The functionalized copolymers of acrylates were applied in various fields such as biomedical [9], textile [10], coating [11], adhesions [12], food packaging [13], and many more [14, 15]. Knowing the copolymer microstructure is useful to predict the properties of the copolymers. The microstructure of copolymers is easily predicted from the values of reactivity ratios of two monomers and the ratios of these values are possible to find from the various linear and non-linear methods [16,17,18].
Polymers are the most preferable host materials to prepare the inorganic nano particles (Nps)-doped composites. These classes of materials have very good applicable properties (i.e., electrical, mechanical, and optical). Materials having both inorganic and organic characteristics were well researched [19,20,21,22,23]. Recently, many techniques [24,25,26,27,28] were developed to prepare the new potential materials that were used in various fields [29,30,31,32,33] such as biological labeling, light-emitting diodes, transistors, solar cells, organic-based electronics, sensors, optical switching, etc.
The numbers of research reports were published on the polymer composites with nano-metal oxides [34, 35], metal sulfides [36, 37], and many more [38,39,40,41]. Among them, semiconducting polymer nano composites of metal sulfides are very useful for their optoelectronic properties. A semiconducting polymer composite of CdS Nps is this class of material. It has broad range of applications in the various fields. The various polymers are used as host materials to prepare the CdS/polymer nano composites. The different CdS-Nps-doped polymer composites are prepared via different techniques. CdS/polystyrene nano-composite was prepared by a chemical route using an ex situ technique [42]. Oxidization polymerization technique was used to prepare CdS/polyaniline nano-composite [43]. CO+2-doped CdS/polyvinyl pyrolidone composite was synthesized by chemical precipitation method [44]. Spin-coating technique was used to prepare thin films of CdS/poly[2-methoxy-5-(2′-ethyl-hexyloxy)-1,4-phenylene vinylene] (MEH-PPV) nano-composite [45]. Several other CdS/polymer nano composites such as CdS/polyamidoamine dendrimer [46], CdS/poly[4-(n-acryloyloxyalkoxy)benzoic acid] [47], and many more [48, 49] were also investigated. Although the properties and synthesis of CdS/polymer nano composites are well investigated, yet the effect of CdS Nps on the degradation of polymer in inert atmosphere (i.e., N2 gas) is not investigated.
The synthesis of macromolecules having antimicrobial properties is useful in various areas like hospitals, dental equipment, water purification systems, food storage, and food packaging units. Polymeric materials having antimicrobial properties are prepared mainly by: (1) polymerization of antimicrobial monomers which have characteristic functional groups responsible for antimicrobial properties and (2) chemical modification of synthetic polymers by antimicrobial agents or biocides. In this article, we have synthesized chlorine containing copolymers and assessed various microorganisms to confirm the copolymer’s antimicrobial potency.
The present paper covers the synthesis, characterization, thermal behavior, and antimicrobial screening of homo- and copolymers of NPEMA with PCPMA. Polymer composites with nano-Cds were prepared and characterized by spectroscopic tools. The main objective of this paper is to study the effect of CdS Nps on thermal degradation of polymer matrix in nano composites.
Experimental
Materials
Ethanol amine, phthalic anhydride, triethyl amine (TEA), N,N-dimethyl formamide (DMF), methanol, cadmium nitrate, sodium sulfide, and 2,2-azo-bis-isobutyronitrile (AIBN) were purchased from the Loba chem Pvt. Ltd. (India). The nutrient broth, potato dextrose broth, and tryptone glucose yeast extract were purchased from the Himedia Laboratories Pvt. Ltd. The pre-grown slants of various microorganisms (i.e., bacteria, yeast, and fungi) were obtained from the Microbial-Type Culture Collection and Gene Bank (MTCC), CSIR-Institute of Microbial Technology, Chandigarh, India. All chemicals are analytical grade and are used as received.
Synthesis
Synthesis of starting materials and monomers
The synthesis of methacryloyl chloride (MAC), N-(2-hydroxy ethyl) phthalimide (NHEP), and 2-(N-phthalimido)ethyl methacrylate (NPEMA) was prepared, as reported in Ref. [50]. The p-chlorophenyl methacrylate (PCPMA) was prepared by earlier reported synthesis [51].
Copolymerization of NPEMA with PCPMA
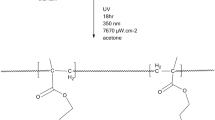
The free-radical polymerization technique [50] was employed to synthesize copolymers of NPEMA with PCPMA having different feed compositions. N,N-Dimethyl formamide (DMF) was used as a solvent and AIBN as a free-radical initiator. The feed composition of monomers is given in Table 1. Appropriate quantities of monomers, DMF, and AIBN (0.5% w/w based on total monomers 1 and 2) were added to a flask fitted with reflux condenser. The reaction mixture was heated to 70 °C for 5 h with stirring. It was kept for cooling at room temperature, and then, the resulting polymer solution was slowly poured in a large volume of methanol with stirring. The polymer is followed out. It was filtered and then washed with methanol. Solid polymers were purified by repeated precipitation from DMF solution using methanol. Synthesis of copolymer is shown in Scheme 1.

Copolymerization (B2–B6) of NPEMA with PCPMA
Preparation of CdS-Nps-doped polymer composites
Polymer (0.5 g) dissolved in 50-ml DMF with stirring in a 250-ml conical flask, and then, the flask was kept for sonication. Under sonication, cadmium nitrate solution (0.1 M) in DMF was added slowly to polymer solution. Then, 0.1-M sodium sulfide (0.11 M) solution prepared in methanol was added dropwise to the solution containing cadmium nitrate and polymer solution under sonication. After complete addition, the solution becomes yellowish and cloudy. The flask is putted for 4 h at room temperature to settle the yellow color polymer composites of nano-CdS. The obtained polymer nano composites were separated by ultracentrifugation. The nano composites were washed several times with methanol to remove unreacted or excess reagents. The polymer nano-composite is dried into a vacuum desiccator.
Characterization
The 1H-NMR resonances were recorded with a Bruckner 400 MHZ FT NMR spectrophotometer. The IR spectra of solid samples in KBr pallets were obtained from Nicolet 6700 FT-IR spectrophotometer. Copolymer compositions and reactivity ratios were determined by spectroscopic data from UV–visible–NIR Schimadzu-3600 spectrophotometer. The phase and crystallographic structures of CdS in polymer nano composites of NPEMA/CMPMA were identified by X-ray diffraction (XRD, Bruckner D2 Phaser 3600 X-ray Diffractometer having Cu-kα radiation source, λ = 0.154 nm). The morphology of polymer nano composites of CdS was derived using transmission electron microscope (Tecnai 20, Philips, Holland, with W-emitter and LaB6 as electron source and accelerating voltage up to 200 kV). Thermal analysis was performed at 10 °C/min heating rate on Mettler-Toledo thermogravimetric analyzer for TGA/DSC-1.
Antimicrobial screening
The quantitative method [50] was used to assess antimicrobial properties. The homo- and copolymers of NPEMA/PCPMA tested against different microorganisms which are commonly employed for biocompatibility test. These microorganisms are bacteria (Escherichia coli, Bacillus subtilis, and Staphylococcus citreus), fungi (Sporotrichum pulverulentum, Aspergillus niger, and Trichoderma lignorum), and yeast (Candida utilis, Pichia stipitis, and Saccharomyces cerevisiae). The antimicrobial activity of poly(NPEMA) was already discussed [50]. All microorganism cultures were prepared from their respected slants. All microorganism cultures were sub-cultured periodically and maintained in their respected media. The bacteria, yeast, and fungi cultures were kept overnight at 37, 30, and 27 °C, respectively. In the present study, the bacterial culture’s %OD is set at 0.1 in 600 nm which corresponds to 108 CFU/ml. The yeast culture’s %OD was set at 0.8 in 600 nm which corresponds to 2 × 106 CFU/ml. The fungi culture was prepared by inoculation with 0.7-mm plug (spore) containing actively growing fungi’s slants. Each test was carried out three times and the mean results are shown in a bar graph.
Results and discussion
1H-NMR spectroscopy
The monomers NPEMA and PCPMA were characterized from the proton NMR spectroscopy. The proton NMR spectra of NPEMA monomer have been discussed in earlier report [50].
NPEMA 1H-NMR(CDCl3) δ (ppm) = 7.5–7.8 (4H, m, Ar–CH), 5.9 (1H, d, vinylic-H), 5.5 (1H, d, vinylic-H), 4.3 (2H, t, –CH2–O), 3.9 (2H, t, –CH2–N), and 1.9 (3H, s, CH3).
PCPMA 1H-NMR(CDCl3) δ (ppm) = 6.9–7.3 (3H, m, Ar–CH), 6.4 (1H, d, vinylic-H), 5.7(1H, d, vinylic-H), 2.3 (3H, s, Ar–CH3), and 2.1 (3H, s, O=C–C–CH3).
FT-IR spectroscopy
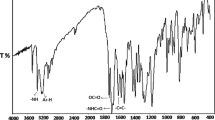
The combined FT-IR spectra (2000–500 cm−1) of homo- and copolymers of NPEMA with PCPMA and their polymer composites with nano-CdS are shown in Figs. 1 and 2, respectively. From the spectra of neat polymers, the bands observed at ~ 1750 cm−1 are assigned for the stretching vibration of –C=O in ester group. Another absorbance observed at ~ 1710 cm−1 corresponds to –C=O stretching in phthalimide [50]. The bands between 1600 and 1400 cm−1 are assigned to the aromatic ring breathing vibration. The same band is also observed in all copolymers IR spectra. Medium bands ~ 1465 and ~ 1387 cm−1 correspond to asymmetrical and symmetrical bending vibration of methyl group, respectively. The bands at 1173, 1146, and 1048 cm−1 are seen in the homopolymers of NPEMA, whereas the bands at 1258, 1200, 1122, 1084, and 1012 cm−1 are seen in the homopolymers of PCPMA which are attributed to the C–C(=O)–O and O–C–C-bending vibration in ester. The bands at 1258, 1199, 1160, 1122, 1080, and 1012 cm−1 are seen in copolymers of NPEMA and PCPMA which are assigned for the bending vibration of C–C(=O)–O and O–C–C in ester. The sharp band at 720 cm−1 is observed in homopolymers of NPEMA. The same absorption band is also observed in the spectra of copolymers and this band’s intensity is decreased with decrease in the NPEMA content in the copolymers. The band ~ 680 cm−1 is assigned for the C–Cl-stretching vibration in homopolymers of PCPMA. The same band is also observed in spectra of copolymers.

FT-IR spectra of poly(NPEMA) (B1); poly(PCPMA) (B7) and nano-Cds-doped polymer composite of poly(NPEMA) (NB1); poly(PCPMA) (NB7)

FT-IR spectra of poly(NPEMA-co-PCPMA) (B2–B6) and nano-Cds-doped polymer composite of poly(NPEMA-co-PCPMA) (NB2–NB6)
In the case of nano-CdS-doped polymer composites, the intensity of the bands is decreased. The intensity of bands corresponding to –C=O (~ 1750 and ~ 1710 cm−1), C–C(=O)–O (1210–1164 cm−1), and O–C–C (1164–1000 cm−1) is effectively decreased and becomes broader. This suggests that the attraction between CdS Nps and polymer matrix occurring on these sites which depict the presence of CdS Nps in polymer matrix affects the vibration of C–O. As NPEMA content decreased in copolymer feed, the interaction between polymer matrix and inorganic matrix also decreased. This was due to the less sites that were available for interactions in the poly(PCPMA) than poly(NPEMA). The band at 1387 cm−1 which corresponds to the symmetric vibration of methyl group is not affected by the presence of CdS Nps. This reveals that no interaction is possible between the methyl and CdS Nps. A weak Van der Waals interaction between polymer matrix and nano-CdS may be envisaged.
Average molecular weights
GPC instrument equipped with Waters 1525 binary pomp, manual injector and connected to Styragel HR 4 column and R.I. detector (waters 2414) is employed to record GPC curves. Tetrahydrofuran (THF) at 1.0 ml/min flow rate is used as a mobile phase throughout the analysis. All the measurements are carried out at 30 °C temperature. About 5–10 mg of each polymer sample is dissolved in 10-ml THF and the resulting solutions are filtered. 20-μl sample solution is injected for the GPC analysis. The average molecular weights, namely, \(\overline{M}\)n, \(\overline{M}\)w, \(\overline{M}\)z, \(\overline{M}\)z + 1, and polydispersity, are shown in Table 2. The GPC data for poly(NPEMA-co-PCPMA) provided the values of \(\overline{M}\)n, \(\overline{M}\)w, \(\overline{M}\)z, and \(\overline{M}\)z + 1 which vary from 20,109 to 22,381, 31,518 to 34,804, 47,784 to 51,883, and 65,954 to 71,200, respectively, for copolymers with different feed ratio of the monomers; polydispersity index lies between 1.54 and 1.60 (Table 2). The results reveal that molecular weights decrease and polydispersity index changes randomly as the NPEMA content decreases in the copolymers. The values of \(\overline{M}\)n, \(\overline{M}\)w, \(\overline{M}\)z, and \(\overline{M}\)z + 1 are, respectively, 26,052, 43,620, 67,474, 91, and 839 for poly(PCPMA) and the polydispersity index is 1.67. The polydispersity of homopolymers and copolymers was nearly 1.5, indicating termination of growing chain by radical combination (Table 2).
X-ray diffraction study and transmission electron microscopy
Powder XRD pattern of nano-CdS-doped poly(NPEMA-co-PCPMA) (0.5/0.5 mol ratio) is shown in Fig. 3. The three peaks are observed in each composite which corresponds to the three reflection planes. These reflection planes [i.e., (111), (220), and (311)] confirm that the CdS Nps are cubic and match with JCPDE number 089-0440. The size of CdS nano particles is calculated using Debye–Scherrer formula [52] for (111) reflection plane of the XRD pattern. The size of CdS Nps varied between 4.11 and 5.32 nm.

XRD pattern of nano-CdS-doped poly(NPEMA-co-PCPMA) composite (mole ratio: 0.5/0.5) (NB4)
Figure 4 shows the TEM images of the nano-CdS-doped poly(NPEMA-PCPMA) (0.5/0.5) (NB4) composite. The images clearly show the formation of CdS nano composites with a spherical shape and homogeneous distribution in polymer matrix.

TEM images of nano-CdS-doped poly(NPEMA-co-PCPMA) composite (mole ratio: 0.5/0.5) (NB4)
Monomer reactivity ratio
The copolymer composition depends on the monomer feed composition and on the relative monomer reactivity. The UV spectroscopy was employed to find the concentration of NPEMA monomer in copolymer. The procedure to find concentration of NPEMA in copolymers using UV spectroscopy was already discussed [50]. The monomer reactivity ratios were determined using four linear methods Fineman–Rose (F–R) [53], inverted Fineman–Rose (Invrt. F–R), Kelan–Tudos (K–T) [54], and extended Kelen–Tudos (Ext. K–T) [55]. The values of r1 (NPEMA), r2 (PCPMA), 1/r1, and 1/r2 and product of r1 and r2 are tabulated in Table 3.
It was seen that the value of r1 is less than r2; this suggests that the reactivity of NPEMA monomer is less than that of PCPMA. This also proves that the PCPMA units were found more in the copolymer content. r1 < r2 suggest that the NPEMA favors the cross propagation, whereas PCPMA favors the homopropagation. As 1/r1 > 1/r2, it was concluded that there were more growing radicals with NPEMA ends than with PCPMA ends due to r1 < r2, which identified which monomer amongst the two was more reactive.
Copolymer microstructure
Mean sequence length
The mean sequence lengths μNPEMA and μPCPMA are calculated using the following equation [56]:
where [M1] and [M2] are concentrations of NPEMA and PCPMA, respectively. The ratio of the mean sequence length distribution μNPEMA/μPCPMA, which theoretically corresponds to the ratio [M1]/[M2], is tabulated in Table 4. For example, at [M1] = 20.0% and [M2] = 80.0%, each copolymer segment with M2 units was approximately six times longer than its adjoining segment with M1 units. The sequence may be expressed as NPPPPPPN, where P stands for PCPMA and N is for NPEMA. The number of NPEMA units in copolymer increases with increasing concentration of NPEMA in the feed. The results of mean sequence length and values of 1/r1 and 1/r2 compare each other very well; 1/r1 is greater than 1/r2, and as expected in copolymers, the homopropagation of PCPMA decreased with decreasing PCPMA in monomer feed, while cross propagation of monomeric units increased with increasing concentration of NPEMA in monomer feed.
Run number and dyad fraction
From the reactivity ratios of monomers, the run number, RN, was determined following Harwood and Reichy [57] as the average number of monomer alternations per 100 monomeric units in a copolymer chain:
The RN value provides a view of sequence distributions in copolymer chains, and consequently, it is possible to assess the relationship between physical properties of copolymers and their compositions. The value of RN can be calculated from reactivity ratios. The variation of RN with X (= [M1]/[M2]) is shown in Fig. 5a.

a Run number for NPEMA and PCPMA monomer pair; b dyad monomer sequence fractions vs. the NPEMA mole fractions for the copolymers NPEMA with PCPMA
The maximum value of RN is 54.2 at X = 1.5 for the poly(NPEMA-co-PCPMA) system. To gain further information about the copolymer structure, the formation probabilities of dyad fractions as a function of the molar fraction of monomeric units in the copolymer can be calculated from the monomer feed compositions and reactivity ratios [58]:
The variation of the dyad fractions with the NPEMA mole fraction in the copolymers is depicted in Fig. 5b. Figure 5b shows S1–1 is less than S2–2, suggesting that NPEMA has negligible attraction with NPEMA monomer, and so, NPEMA homopolymerization is not favorable. The value of S1–2 implied that homopolymerization of PCPMA is quite dominant. The consequences of these calculations are the same as those obtained from reactivity ratios.
Thermal analysis
Thermal stability of polymer and their composites with nano-CdS
Thermogravimetric analysis (TGA) technique was used to investigate the thermal stability of polymers and their composite with nano-CdS. TGA traces of homo- (B1 and B7) and copolymers of NPEMA with PCPMA (B2–B6) and its nano composites (NB1–NB7) are shown in Figs. 6 and 7, respectively. The % weight loss at various temperatures, decomposition range, Tmax, T50, and integral decomposition temperature (IPDT) for homo- and copolymers of NPEMA with PCPMA are tabulated in Table 5. The TGA traces show that the decomposition of polymers occurs in two steps. The first decomposition step was in the range of 165–397 °C, whereas the range between 314 and 530 °C was observed for second decomposition step. The first step seen at lower temperature range corresponds to the smaller polymeric chain degradation. The second step observed at higher range corresponds to the scission of main polymeric chain. Poly(PCPMA) has higher thermal decomposition steps than other polymers which confirms that the thermal degradation of poly(PCPMA) was slower. It was also notable (from Table 5) that the % weight loss of poly(PCPMA) at 700 °C was slightly lower than the poly(NPEMA), whereas the % weight loss of copolymers (B2–B6) was relatively higher than each other. The IPDT of homopolymers of NPEMA and PCPMA is, respectively, 351 and 467 °C. The IPDT of copolymers of NPEMA with PCPMA varied between 333 and 347 °C. It was concluded that with the increase of PCPMA feed in copolymers, the thermal stability of copolymers (B2–B6) was also increased. Overall, the neat polymers and polymer nano composites show the two-step thermal degradation.

TGA traces of neat homo- (B1 and B7) and copolymers (B2–B6) of NPEMA/PCPMA

TGA traces of nano-CdS-doped homo- (NB1 and NB7) and copolymers (NB2–NB6) of NPEMA/PCPMA
Kinetic and thermodynamic parameters of the thermal degradation
The stability of polymers and their composites with nano-CdS was also confirmed from the kinetic (i.e., Ea) and thermodynamic (i.e., ΔH, ΔS, and ΔG) parameters of the thermal degradation of respective polymers and polymer nano composites. The kinetic parameters of thermal degradation were obtained using Briodo (BR) [59] and Coats–Redfern (CR) [60] methods. The activation energy (by BR) was evaluated from the slope of the ln [ln (1/y)] vs. 1/T plot, whereas activation energy (by CR) was obtained from the slope of ln [− ln (1 − y)/T2] vs. 1/T plot. In these entire calculations. y = (Wt − Wα)/(W0 − Wα), where Wt is the weight at temperature, W0 is initial weight, and Wα is the weight at the end of pyrolysis.
Activation energies (in kJ/mol) for homopolymers of NPEMA and PCPMA for both degradation steps obtained from Broido method were, respectively (56 and 83 kJ/mol) and (80 and 145 kJ/mol); the same from Coats–Redfern were (56 and 80 kJ/mol) and (85 and 150 kJ/mol). The energy of activation in copolymers B2 (80:20; NPEMA:PCPMA) obtained from Broido was (50 and 69 kJ/mol) and Coats–Redfern provided the same values. The energies of activation for other copolymers (B3–B7) with different monomer feed are shown in Table 6. It was noticeable that the activation energy of the first degradation step was lower than that of the second step. This concludes that less energy is required for degradation of shorter polymeric chains, whereas higher activation energy corresponds to the higher energy needed for degradation of higher polymeric chains. The activation energy of poly(PCPMA) was higher than that of poly(NPEMA). The activation energies of copolymers (B2–B6) randomly increased or decreased with increase in PCPMA content in copolymer feed.
Thermodynamic parameters (i.e., ∆S, ∆H, and ∆G) of thermal degradation were also useful to confirm the thermal stability of polymers. The value of enthalpy change (∆H) was positive, which indicated the endothermic nature of thermal degradation of polymers. Lower the value of ∆H, lower the difference between potential energy barrier of reagents and activated complex. This suggests that the formation of activated complex was easily favored, and hence, degradation process was faster [61, 62]. How close the system to its equilibrium could define from the value of entropy change (ΔS). Lower the value of ΔS suggests that the formation of activated complex was slow and vice versa [61, 62]. The negative value of ∆S and positive value of ∆G for a decomposition process indicate that steps are non-spontaneous. As the values of ∆G increase, the process of formation of activated complex was slow which means that thermal degradation process was slower and vice versa.
The values of thermodynamic parameters for neat polymers indicate that poly(NPEMA) (B1) was less stable than the poly(PCPMA) (B7) and poly(NPEMA-co-PCPMA) (B2–B6). It was notified from Table 6, and the first decomposition step has the lowest values of Ea, ∆H, and ∆G than the second decomposition step. This reveals that the first decomposition step was faster than the second decomposition step. This was further supported from the higher value of ∆S for first decomposition step.
The obtained values of kinetic and thermodynamic parameters for polymer composites with nano-CdS are tabulated in Table 7. The values of the activation energy obtained from both Broido and Coats–Redfern were found to be same. The values of activation energies for both steps of degradation for polymer composites of poly(NPEMA) and poly(PCPMA) doped with nano-CdS were 28 and 30 and 27 and 83 kJ/mol, respectively. The values of activation energies for both degradation steps of copolymer composites of poly(NPEMA-co-PCPMA) with nano-CdS varied between 20–34 and 30–78 kJ/mol, respectively. The values of activation energy were found relatively lower for polymer nano composites (NB1–NB7) than the neat polymers (B1–B7). It was also noticed that the values of ∆H and ∆G for polymer composites were found to be relatively lower than their corresponding neat polymers. In case of ∆S, the values were found higher than the neat polymers. Overall, the result reveals that the decomposition of polymer matrix in polymer nano composites was easily favorable than in neat polymers. It was also concluded that the neat polymers were relatively more stable than their respective polymer nano composites. In case of polymer nano composites, the inorganic nano particles can affect the structure of polymers. These result in higher or lower thermal stability of polymer nano composites. Few research papers were found in this area (Table 8). The neat polyamide 6, polyethylene oxide (PEO)/polyvinyl alcohol (PVA), isotactic polypropylene (iPP), and poly(butylene succinate-co-adipate) (PBSA) have 175, 101, 220.57, and 159.9 kJ/mol of activation energies, respectively. The activation energies of their nano composites, respectively, with clay, ZnO, palladium (Pd), and organically modified synthetic fluorine mica were higher than neat polymers. These suggest that the stronger interaction between polymer matrix and inorganic nano particles than the polymer nano composites has higher value of activation energy than the corresponding neat polymers [63, 66,67,68]. The activation energies of neat poly(3-hydroxy butyrate) (PHB), polyvinyl ester and nylon 6 were, respectively, 136.8, 186.3, and 276 kJ/mol, while the activation energies of their nano-composite, respectively, with Ag2S, O-montmorillonite (O-MMT), and Glass fibers were found lower than neat polymers. This is due to the weak interaction between polymer matrix and inorganic nano particles [64, 65]. The lower value of kinetic and thermodynamic parameters suggests the catalytic effect arises in polymer nano composites.
Antimicrobial screening
The functional polymers having chlorine and fluorine groups were mostly used for antimicrobial properties. The effectiveness of homo- and copolymers of NPEMA and PCPMA to hinder the growth of microorganisms is shown in Fig. 8. It appears that compared to poly(NPEMA) and poly(NPEMA-co-PCPMA), the poly(PCPMA) more effectively hinders the microorganism’s growth. The copolymers (Fig. 8a) allowed growth of 30–20, 32–25, and 50–38% for E. coli, B. subtilis, and S. citreus, respectively. The poly(NPEMA) tolerated, respectively, 35, 42, and 52% growth for E. coli, B. subtilis, and S. citreus; poly(PCPMA) allowed, respectively, 10, 18, and 20% growth for the species indicated above.
The effect of homo- and copolymers of NPEMA and PCPMA on the growth of different fungi is demonstrated in Fig. 8b. It is seen that the hindrance to growth by poly(NPEMA) was less than their corresponding copolymers with PCPMA. The effect of poly(NPEMA) on the fungi has already been indicated in the preceding section. Growths of A. niger, S. pulverulentum, and T. lignorum, respectively, were 30, 20, and 34% in poly(PCPMA). The copolymers allowed 52–28, 48–28, and 58–40% growth of A. niger, S. pulverulentum, and T. lignorum, respectively.
The poly(NPEMA-co-PCPMA) and poly(PCPMA) were more effective than the poly(NPEMA) to reduce the growth of yeast (Fig. 8c). The poly(NPEMA) permitted 40, 50, and 58% growth of S. cerevisiae, C. utilis and P. stipitis, respectively; poly(PCPMA) allowed, respectively, 18, 32, and 28% growth of the same. The copolymers tolerated 38–22, 50–38, and 56–40% growth of S. cerevisiae, C. utilis, and P. stipitis, respectively.
In general, poly(PCPMA) hindered the growth of all the microorganisms effectively, while the poly(NPEMA) was less effective than poly(PCPMA) and their copolymers. This may be traced to the presence of chlorine in poly(PCPMA) and poly(NPEMA-co-PCPMA). It was observed that increasing the concentration of PCPMA in copolymers resulted in increase of antimicrobial properties and vice versa. It was further seen that the homo- and copolymers of PCPMA effectively hindered the growth of bacteria, while the growth of yeast and fungi was moderately hindered.

Antimicrobial properties of homo- and copolymers of NPEMA/PCPMA—a bacteria; b fungi, and c yeast
Overall, the percentage of NPEMA in the copolymers increased, the effectiveness of the copolymers to inhibit the growth of microorganisms decreased, while increase in the percentage of monomers which have active groups (i.e., –Cl) will increase the ability of polymers to inhibit the growth of microorganisms. This investigation indicates that monomers containing chlorine can be used to prepare polymers which can be used as general antimicrobial agents in water purification systems and food packaging materials.
Conclusion
The homo- and copolymers (with different monomer feeds) of NPEMA and PCPMA were prepared via free radical solution polymerization technique. The relative intensity of different bands (i.e., C=O group, –CH2 rocking vibration) in FT-IR spectra of copolymers was increased or decreased. This reveals that the composition of monomers in copolymers was well matched. The polydispersity of homo- and copolymers varied between 1.45 and 1.67. Nano-CdS-doped polymer composite was prepared via in situ technique. The nano-CdS-doped polymer composite is characterized by XRD and FT-IR. The XRD pattern confirms the cubic structure of CdS nano particles with three reflection planes (111), (222), and (311). The FT-IR spectra of polymer nano composites confirm the weak interaction between inorganic nano particles and polymer matrix. The TGA analysis of polymers confirms that the poly(PCPMA) was more stable than poly(NPEMA-co-PCPMA) and poly(NPEMA). The values of activation energy, ∆S, ∆H, and ∆G, confirm the stability of poly(PCPMA). The values of activation energy for polymer nano composites were found lower than the neat polymers. This confirms the lower thermal stability of polymer nano composites than the neat polymers. The lower thermal stability of polymer nano composites also confirms from the values of ∆S, ∆H, and ∆G. It is possible that CdS nano particles act as a catalyst during the degradation of polymer nano composites.
References
Atabaki F, Abdolmaleki A, Barati A (2016) Free radical copolymerization of methyl methacrylate and N-2-methyl-4-nitro-phenylmaleimide: improvement in the T g of PMMA. Colloid Polym Sci 294:455–462. https://doi.org/10.1007/s00396-015-3799-x
Fan L, Zhao D, Bian C et al (2011) Glass transition temperatures of copolymers from methyl methacrylate, styrene, and acrylonitrile: binary copolymers. Polym Bull 67:1311–1323. https://doi.org/10.1007/s00289-011-0528-6
Chee S, Lee MK, Lee CL (2016) Thermal properties of poly(methyl methacrylateco-butyl acrylate-co-acrylic acid) modified with divinyl benzene and vinyl trimethoxy silane. J Macromol Sci PART A 53:82–87. https://doi.org/10.1080/10601325.2016.1120175
Penfold HV, Holder SJ, Kenzie BEM (2010) Octadecyl acrylate–Methyl methacrylate block and gradient copolymers from ATRP: comb-like stabilizers for the preparation of micro- and nano-particles of poly (methyl methacrylate) and poly (acrylonitrile) by non-aqueous dispersion polymerization. Polymer (Guildf) 51:1904–1913. https://doi.org/10.1016/j.polymer.2010.02.029
Kulikov EE, Zaitsev SD, Semchikov YD (2015) Reversible addition fragmentation chain transfer (RAFT) polymerization of isobornyl acrylate. Polym Sci Ser C 57:120–127. https://doi.org/10.1134/S1811238215010051
Wang X, Sun X, Zhai G (2015) Aqueous radical polymerization of N,N-dimethylacrylamide redox-initiated by aerobically catalytic oxidation of water-soluble tertiary amines. Polym Bull 72:2809–2829. https://doi.org/10.1007/s00289-015-1437-x
Kumar D, Jaikumar V (2015) Synthesis, characterization and kinetic study of photocrosslinkable prepolymer: 2-hydroxy-3-[p-(1-{p-[2-hydroxy-3-(vinylcarbonyloxy)propoxy]phenol}-1-phenylethyl)phenoxy]propylacrylate. Des Monomers Polym 18:295–302. https://doi.org/10.1080/15685551.2014.999469
Yao W, Li Y, Huang X (2014) Fluorinated poly (meth) acrylate: synthesis and properties. Polymer (Guildf). 55:6197–6211. https://doi.org/10.1016/j.polymer.2014.09.036
Zho K, Lin B, Cui W, Feng L, Chen T, Wei J (2014) Preparation and adsorption of bovine serum albumin-imprinted polyacrylamide hydrogel membrane grafted on non-woven polypropylene. Talanta 121:256–262
Najafi H, Yazdanshenas EM, Rashidi A, Montazer M (2009) Synthesis and characterization of styrene-acrylic binders and their application on pigment printing of cotton and polyester textile fabrics. Asian J Chem 21:4871–4880
Kim D, Park HJ, Lee KY (2015) Study on curing behaviors of epoxy acrylates by UV with and without aromatic component. Macromol Res 23:944–951. https://doi.org/10.1007/s13233-015-3130-1
Mageswari S, Subramanian K (2012) Synthesis, characterization, and study of antibacterial activity of homopolymers and copolymers of 4-benzyloxyphenylacrylates for pressure-sensitive adhesive application. J Appl Polym Sci 125:315–3124. https://doi.org/10.1002/app
Suppakul P, Miltz J, Sonneveld K, Bigger SW (2003) Active packaging technologies with an emphasis on antimicrobial concise reviews in food science. J Food Sci 68:408–420
Zhao K, Chen T, Lin B, Cui W, Kan B, Yang N, Zhou X, Zhang XX, Wei J (2015) Adsorption and recognition of protein molecular imprinted calcium alginate/polyacrylamide hydrogel film with good regeneration performance and high toughness. React Funct Polym 87:7–14
Zhang X, Lin B, Zhao K, Wei J, Guo J, Cui W, Jiang S, Yang Y, Li J (2015) A freestanding calcium alginate/polyacrylamide hydrogel nanofiltration membrane with high anti-fouling performance: preparation and characterization. Desalination 365:234–241
Lerari D (2015) Synthesis and characterization of new copolymer based cinnamyl methacrylate monomer: determination of monomer reactivity ratio and statistical sequence 2 Experimental Part. Macromol Res 18:1008–1014
Shekh MI, Patel KP, Patel RM (2015) Copolymers of phthalimide moiety containing 2-(N-phthalimido) ethyl methacrylate (NPEMA) with p-acetamidophenyl methacrylate (PAPMA):synthesis, characterization, thermal properties and antimicrobial activity. J Chem Pharm Res 7:358–367
Patel DM, Shekh MI, Patel KP, Patel RM (2015) Synthesis, characterization and antimicrobial activity of novel acrylic materials. J Chem Pharm Res 7:470–480
Saleh TA, Gupta VK (2012) Photo-catalyzed degradation of hazardous dye methyl orange by use of a composite catalyst consisting of multi-walled carbon nanotubes and titanium dioxide. J Colloid Interface Sci 371:101–106. https://doi.org/10.1016/j.jcis.2011.12.038
Sahiner N (2006) In situ metal particle preparation in cross-linked poly (2-acrylamido-2-methyl-1-propansulfonic acid) hydrogel networks. Colloid Polym 285:283–292. https://doi.org/10.1007/s00396-006-1562-z
Wei S, Zhao K, Zhang X, Fu Y, Li Z, Xu S, Wei J (2015) Adsorption and photocatalytic degradation of dyes on polyacrylamide/calcium alginate/TiO2 composite film. Functional Materials Letters 8:1–5
Gupta VK, Saleh TA (2013) Sorption of pollutants by porous carbon, carbon nanotubes and fullerene—an overview. Environ Sci Pollut Res 20:2828–2843. https://doi.org/10.1007/s11356-013-1524-1
Gupta VK, Sharma S (2003) Removal of zinc from aqueous solutions using bagasse fly ash—a low cost adsorbent. Ind Eng Chem Res 42:6619–6624
Saravanan R, Gupta VK, Prakash T, Narayanan V, Stephen A (2013) Synthesis, characterization and photocatalytic activity of novel Hg doped ZnO nanorods prepared by thermal decomposition method. J Mol Liq 178:88–93. https://doi.org/10.1016/j.molliq.2012.11.012
Sahiner N, Sel K, Meral K, Onganer Y, Butun S, Ozay O, Silan C (2011) Colloids and Surfaces A: physicochemical and engineering aspects hydrogel templated CdS quantum dots synthesis and their characterization. Colloids Surf A Physicochem Eng Asp 389:6–11. https://doi.org/10.1016/j.colsurfa.2011.09.006
Saravanan R, Khan MM, Gupta VK, Vargas E, Gracia F, Arumainathan S, Stephen A (2015) ZnO/Ag/Mn2O3 nanocomposite for visible light-induced industrial textile effluent degradation, uric acid and ascorbic acid sensing and antimicrobial activities. RSC Adv 5:3464534651. https://doi.org/10.1039/C5RA02557E
Rajendran S, Khan MM, Gracia F, Qin J, Gupta VK, Stephen A (2016) Ce3+-ion-induced visible-light photocatalytic degradation and electrochemical activity of ZnO/CeO2 nanocomposite. Sci Rep. https://doi.org/10.1038/srep31641
Saravanan R, Sacari E, Gracia F, Khan M, Mosquera M, Gupta VK (2016) Conducting PANI stimulated ZnO system for visible light photo catalytic degradation of coloured dyes. J Mol Liq 221:1029–1033. https://doi.org/10.1016/j.molliq.2016.06.074
Saleh TA, Gupta VK (2012) Photo-catalyzed degradation of hazardous dye methyl orange by use of a composite catalyst consisting of multi-walled carbon nanotubes and titanium dioxide. J Colloid Interface Sci 371:101–106. https://doi.org/10.1016/j.jcis.2011.12.038
Devaraj M, Saravanan R, Deivasigamani R et al (2016) Fabrication of novel shape Cu and Cu/Cu2O nanoparticles modified electrode for the determination of dopamine and paracetamol. J Mol Liq 221:930–941. https://doi.org/10.1016/j.molliq.2016.06.028
Khani H, Kazem M, Arab P, Gupta VK, Vafaei Z (2010) Multi-walled carbon nanotubes-ionic liquid-carbon paste electrode as a super selectivity sensor: application to potentiometric monitoring of mercury ion (II). J Hazard Mater 183:402–409. https://doi.org/10.1016/j.jhazmat.2010.07.039
Arya SK, Dey A, Bhansali S (2011) Biosensors and Bioelectronics Polyaniline protected gold nanoparticles based mediator and label free electrochemical cortisol biosensor. Biosens Bioelectron 28:166–173. https://doi.org/10.1016/j.bios.2011.07.015
Kim WH, Mäkinen AJ, Nikolov N et al (2002) Molecular organic light-emitting diodes using highly conducting polymers as anodes Molecular organic light-emitting diodes using highly conducting polymers as anodes. Appl Phys Lett 80:3844. https://doi.org/10.1063/1.1480100
Gupta VK, Jain R, Nayak A, Agarwal S, Shrivastava M (2011) Removal of the hazardous dye—Tartrazine by photodegradation on titanium dioxide surface. Mater Sci Eng C 31:1062–1067. https://doi.org/10.1016/j.msec.2011.03.006
Gupta VK, Nayak A (2012) Cadmium removal and recovery from aqueous solutions by novel adsorbents prepared from orange peel and Fe2O3 nanoparticles. Chem Eng J 180:81–90. https://doi.org/10.1016/j.cej.2011.11.006
Asfaram A, Ghaedi M, Agarwal S, Tyagi I, Gupta VK (2015) Removal of basic dye Auramine-O by ZnS: Cu nanoparticles loaded on activated carbon Optimization of parameters using response surface methodology with central composite design. RSC Adv 5:18438–18450. https://doi.org/10.1039/C4RA15637D
Farooq M, Sagbas S, Yildiz M, Meral K, Siddiq M, Aktas N, Sahiner N (2017) Gum arabic microgels as template for in situ metal-sulfide based quantum dots preparation and their thermal, spectroscopic, optical, and magnetic characterization. J Electron Mater 46:4373–4383. https://doi.org/10.1007/s11664-017-5394-z
Saravanan R, Gupta VK, Narayanan V, Stephen A (2014) Visible light degradation of textile effluent using novel catalyst ZnO/γ-Mn2O3. J Taiwan Inst Chem Eng 45:19101917. https://doi.org/10.1016/j.jtice.2013.12.021
Saravanan R, Gracia F, Mansoob M, Poornima V, Gupta VK, Narayanan V, Stephen A (2015) ZnO/CdO nanocomposites for textile effluent degradation and electrochemical detection. J Mol Liq 209:374–380. https://doi.org/10.1016/j.molliq.2015.05.040
Sahiner N, Sagbas S, Yoshida H, Lyon LA (2014) Synthesis and properties of inulin based microgels. Colloids Interface Sci Commun 2:15–18. https://doi.org/10.1016/j.colcom.2014.08.003
Saravanan R, Joicy S, Gupta VK et al (2013) Visible light induced degradation of methylene blue using CeO2/V2O5 and CeO2/CuO catalysts. Mater Sci Eng C 33:4725–4731. https://doi.org/10.1016/j.msec.2013.07.034
Tripathi SK, Kaur R (2015) Investigation of non-linear optical properties of CdS/PS polymer nanocomposite synthesized by chemical route. Opt Commun 352:55–62. https://doi.org/10.1016/j.optcom.2015.04.042
Kose TD, Ramteke SP (2012) Studies on synthesis and electrical properties of CdS-polyaniline nanocomposite via oxidation polymerization. Int J Compos Mater 2:44–47. https://doi.org/10.5923/j.cmaterials.20120204.01
Muruganandam S, Anbalagan G, Murugadoss G (2015) Optical, electrochemical and thermal properties of CO2+ -doped CdS nanoparticles using poly(vinylpyrrolidone). Appl Nano sci 5:245–253. https://doi.org/10.1007/s13204-014-0313-6
Laera AM, Resta V, Piscopiello E, Miceli V, Schioppa M, Scalone AG, Benedetto F, Tapfer L (2013) In situ growth of well-dispersed CdS nanocrystals in semiconducting polymers. Nanoscale Res Lett 8:382
Wu XC, Bittner AM, Kern K (2005) Synthesis, photoluminescence, and adsorption of CdS/dendrimer nanocomposites. J Phys Chem B 109:230–239
Ezhov AA, Shandryuk GA, Bondarenko GN, Merekalov AS, Abramchuk SS, Shatalova AM, Manna P, Zubarev ER, Talroze RV (2011) Liquid-crystalline polymer composites with CdS nanorods: structure and optical properties. Langmuir 27:13353–13360
Yazdani S, Hatami M, Vahdat SM (2014) The chemistry concerned with the sonochemical-assisted synthesis of CeO2/poly (amic acid) nanocomposites. Turkish J Chem 38:388–401. https://doi.org/10.3906/kim-1306-33
Basu S, Mondal S, Chatterjee U, Mandal D (2009) Poly (styrene-b-2-(N,N-dimethylamino) ethyl methacrylate) diblock copolymers: micellization and application in the synthesis of photoluminescent CdS nanoparticles. Mater Chem Phys 116:578–585. https://doi.org/10.1016/j.matchemphys.2009.04.040
Shekh MI, Patel DM, Patel KP, Patel RM (2016) Electrospun nanofibers of Poly(NPEMA-co-CMPMA): used as heavy metal ion remover and water sanitizer. Fibers Polym 17:358–370. https://doi.org/10.1007/s12221-016-5861-9
Patel MV, Patel JN, Ray A, Patel RM (2005) Synthesis, characterization, thermal properties, and antimicrobial activity of 4-chloro-3-methyl phenyl methacrylate/8-quinolinyl methacrylate copolymers. J Polym Sci Part A Polym Chem 43:157–167. https://doi.org/10.1002/pola.20488
Narayandass SK, Kumar R, Mangalaraj D, Narayandass SK, Kim K, Yi J (2004) Optoelectronic properties of Zn0.52Se0.48/Si Schottky diodes. Solid State Electron 48:2219–2223. https://doi.org/10.1016/j.sse.2004.05.082
Fineman M, Ross SD (1948) Linear method for determining monomer reactivity ratios in copolymerization. J Polym Sci V:259–262
Tüdos F, Kelen T, Foldes-Berezsnich T, Turcsanyi B (1976) Analysis of linear methods for determining copolymerization reactivity ratios. III. Linear graphic method for evaluating data obtained at high conversion levels. J MacromolSci Part A Chem Pure Appl Chem A10:1513–1540. https://doi.org/10.1080/00222337608060768
Kelen T, Tudos F (1875) Analysis of the linear methods for determining copolymerization reactivity ratios. I. A new improved linear graphic method. J Macromol Sci Part A- Chem Pure Appl Chem A9:1–27
Arshady R, Kenner GW, Laboratories RR (1974) Phenolic resins for solid-phase peptide synthesis: copolymerization of styrene and p-Acetoxystyrene. J Polym Sci 12:2017–2025
Harwood HJ, Richey WM (1964) The characterization of sequence distribution. J Polym Res Part-B Polym Lett 2:601–607
Ignarshi S (1963) Representation of composition and blockiness of the copolymer by a triangular coordinate system. J Polym Sci Part B Polym Lett 1:359–363
Broido A (1969) A simple, sensitive graphical method of treating thermogravimetric analysis data. J Polym Sci Part A-2 Polym Phys 1773:1761–1773. https://doi.org/10.1002/pol.1969.160071012
Coats W, Redfern JP (1964) Kinetic parameters from thermogravimetric data. Nature 201:68–69
Georgieva V, Zvezdova D, Vlaev L (2012) Non-isothermal kinetics of thermal degradation of chitosan. Chem Cent J 2012:681–690
Georgieva V, Zvezdova D, Vlaev L (2013) Non-isothermal kinetics of thermal degradation of chitin. J Therm Anal Calorim 111:763. https://doi.org/10.1007/s10973-012-2359-6
Pramoda KP, Liu T, Liu Z et al (2003) Thermal degradation behavior of polyamide 6/clay nanocomposites. Polym Degrad Stab 81:47–56. https://doi.org/10.1016/S0141-3910(03)00061-2
Yeo SY, Tan WL, Bakar MA, Ismail J (2010) Silver sulfide/poly (3-hydroxybutyrate) nanocomposites: thermal stability and kinetic analysis of thermal degradation. Polym Degrad Stab 95:1299–1304. https://doi.org/10.1016/j.polymdegradstab.2010.02.025
Pashaei S, Syed AA, Pashaei S, Syed AA (2011) Thermal characteristics of nanostructured filler-incorporated Poly (vinyl ester) nanocomposites. Polym Plast Technol Eng 50:973–982. https://doi.org/10.1080/03602559.2010.551378
El-kader FHA, Hakeem NA, Elashmawi IS, Ismail AM (2013) Structural, optical and thermal characterization of ZnO nanoparticles doped in PEO/PVA blend films. Aust J Basic ApplSci 7:608–619
Lee YJ, Lee H, Hong S, Choi I (2012) Thermal Degradation kinetics of iPP/Pd nanocomposite prepared by a drying process. Adv Mater PhysChem 2:110–114. https://doi.org/10.4236/ampc.2012.24B030
Ray SS, Bandyopadhyay J, Bousmina M (2007) Thermal and thermomechanical properties of poly(Butylene succinate)-co-Adipate] nanocomposite. PolymDegrad Stab 92:802–812. https://doi.org/10.1016/j.polymdegradstab.2007.02.002
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shekh, M.I., Patel, D.M., Patel, N.N. et al. Methacrylate copolymers and their composites with nano-CdS: synthesis, characterization, thermal behavior, and antimicrobial properties. Int J Ind Chem 9, 153–166 (2018). https://doi.org/10.1007/s40090-018-0146-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-018-0146-3