
Abstract
The oculocerebrorenal disorder of Lowe syndrome is an X-linked mutation in the gene oculocerebrorenal syndrome of Lowe 1 (OCRL), characterized by the triad of congenital cataracts, severe intellectual impairment, and renal tubular dysfunction. Manifestations of phenotype in female carriers and patients are extremely rare. We present a female case with congenital cataracts, severe intellectual impairment, sensorineural hearing loss, and renal tubular dysfunction as Lowe syndrome. A 9-year-old Japanese girl visited our hospital due to prolonged proteinuria. Her renal biopsy revealed diffuse mesangium proliferation, sclerosis and dilatation of renal tubules, and mild IgA deposition in the mesangial region. Furthermore, she had congenital cataracts, severe intellectual impairment, and sensorineural hearing loss. Genetic screening did not identify mutations of the ORCL gene encoding inositol polyphosphate 5-phosphatase (IPP-5P) (46 XX, female). However, we found the reduction of enzyme activity of IPP-5P to 50% of the normal value. Furthermore, her renal function had deteriorated to renal failure within a decade. Finally, she received peritoneal dialysis and renal transplantation. We present the oculocerebrorenal phenotype of Lowe syndrome in a female patient with reduced activity of IPP-5P without OCRL gene mutation.

Similar content being viewed by others
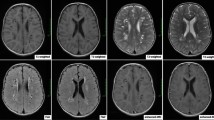
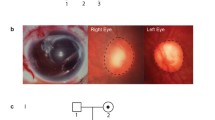
Availability of data and materials
Data and materials are available from our hospital.
References
Lowe CU, Terrey M, MacLachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. Am J Dis Child. 1952;83(2):164–84.
Bökenkamp A, Ludwig M. The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol. 2016;31(12):2201–12.
Hichri H, Rendu J, Monnier N, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat. 2011;32(4):379–88.
Recker F, Reutter H, Ludwig M. Lowe syndrome/Dent-2 disease: a comprehensive review of known and novel aspects. J Pediatr Genet. 2013;2(2):53–68.
Uemura O, Nagai T, Ishikura K, et al. Creatinine-based equation to estimate the glomerular filtration rate in Japanese children and adolescents with chronic kidney disease. Clin Exp Nephrol. 2014;18:626–33.
Sekine T, Nozu K, Iyengar R, et al. OCRL1 mutations in patients with Dent disease phenotype in Japan. Pediatr Nephrol. 2007;22(7):975–80.
Zhang X, Jefferson AB, Auethavekiat V, Majerus PW. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci USA. 1995;92(11):4853–6.
Svorc JM, Masopust J, Komárková A, Macek M, Hyánek J. Oculocerebrorenal syndrome in a female child. Am J Dis Child. 1967;114(2):186–90.
Harris LS, Gitter KA, Galin MA, Plechaty GP. Oculo-cerebro-renal syndrome. Report of a case in a baby girl. Br J Ophthalmol. 1970;54(4):278–80.
Hodgson SV, Heckmatt JZ, Hughes E, Crolla JA, Dubowitz V, Bobrow M. A balanced de novo X/autosome translocation in a girl with manifestations of Lowe syndrome. Am J Med Genet. 1986;23(3):837–47.
Mueller OT, Hartsfield JK, Gallardo LA, et al. Lowe oculocerebrorenal syndrome in a female with a balanced X;20 translocation: mapping of the X chromosome breakpoint. Am J Hum Genet. 1991;49(4):804–10.
Cau M, Addis M, Congiu R, et al. A locus for familial skewed X chromosome inactivation maps to chromosome Xq25 in a family with a female manifesting Lowe syndrome. J Hum Genet. 2006;51(11):1030–6.
Pirruccello M, De Camilli P. Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem Sci. 2012;37(4):134–43.
Suchy SF, Olivos-Glander IM, Nussabaum RL. Lowe syndrome, a deficiency of phosphatidylinositol 4,5-bisphosphate 5-phosphatase in the Golgi apparatus. Hum Mol Genet. 1995;4(12):2245–50.
Wu G, Zhang W, Na T, Jing H, Wu H, Peng JB. Suppression of intestinal calcium entry channel TRPV6 by OCRL, a lipid phosphatase associated with Lowe syndrome and Dent disease. Am J Physiol Cell Physiol. 2012;302(10):C1479–1491.
Monserrat L, Gimeno-Blanes JR, Marín F, et al. Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50(25):2399–403.
Abbassi V, Lowe CU, Calcagno PL. Oculo-cerebro-renal syndrome. A review. Am J Dis Child. 1968;115(2):145–68.
Tricot L, Yahiaoui Y, Teixeira L, et al. End-stage renal failure in Lowe syndrome. Nephrol Dial Transplant. 2003;18(9):1923–5.
Witzleben CL, Schoen EJ, Tu WH, McDonald LW. Progressive morphologic renal changes in the oculo-cerebro-renal syndrome of Lowe. Am J Med. 1968;44(2):319–24.
Nakanishi K, Yoshikawa N. Immunoglobulin a nephropathies in children (includes HSP). In: Avner ED, Harmon WE, Niaudet P, editors. Pediatric nephrology. Springer: Heidelberg; 2016. p. 983–1033.
Schena FP, Cerullo G, Rossini M, Lanzilotta SG, D’Altri C, Manno C. Increased risk of end-stage renal disease in familial IgA nephropathy. J Am Soc Nephrol. 2002;13(2):453–60.
Hodgson SV, Heckmatt JZ, Hughes E, Crolla JA, Dubowitz V, Bobrow M. A balanced de novo X/autosome translocation in a girl with manifestations of Lowe syndrome. Am J med Genet. 1986;23:837–47.
Acknowledgements
We are grateful to the past Dr. Youichi Mizusawa at Tokyo Medical and Dental University for enzyme assays and the past Dr. Takashi Sekine at The University of Tokyo for genetic investigations. We also thank Dr. Makoto Ishida at Ishida Clinic and Dr. Hidehito Kondou at Japanese Red Cross Kyoto Daiichi Hospital for useful discussions and Dr. Mai Ihashi, other pediatricians, and many medical staff working in Minoh City Hospital for their clinical co-operation.
Funding
This research was not supported by any funds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Informed consent was obtained from patient and her parent included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Yamamoto, K., Hasegawa, Y., Ohata, Y. et al. Complete oculocerebrorenal phenotype of Lowe syndrome in a female patient with half reduction of inositol polyphosphate 5-phosphatase. CEN Case Rep 9, 95–100 (2020). https://doi.org/10.1007/s13730-019-00434-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-019-00434-z