
Abstract
An outbreak of a cluster of viral pneumonia cases, subsequently identified as coronavirus disease 2019 (COVID-19), due to a novel SARS-CoV-2 necessitates an urgent need for a vaccine to prevent infection or an approved medication for a cure. In our in silico molecular docking study, a total of 173 compounds, including FDA-approved antiviral drugs, with good ADME descriptors, and some other nucleotide analogues were screened. The results show that these compounds demonstrate strong binding affinity for the residues at the active sites of RNA-dependent RNA-polymerase (RdRp) modelled structures and Chymotrypsin-like cysteine protease (3CLpro) of the HCoV proteins. Free energies (ΔG’s) of binding for SARS-CoV-2 and SARS-CoV RdRp range from – 5.4 to – 8.8 kcal/mol and – 4.9 to – 8.7 kcal/mol, respectively. Also, SARS-CoV-2 and SARS-CoV 3CLpro gave ΔG values ranging from − 5.1 to − 8.4 kcal/mol and − 5.5 to − 8.6 kcal/mol, respectively. Interesting results are obtained for ivermectin, an antiparasitic agent with broad spectrum activity, which gave the highest binding energy value (− 8.8 kcal/mol) against the 3CLpro of SARS-CoV-2 and RdRps of both SARS-CoV and SARS-CoV-2. The reason for such high binding energy values is probably due to the presence of hydroxy, methoxy and sugar moieties in its structure. The stability of the protein–ligand complexes of polymerase inhibitors considered in this investigation, such as Sofosbuvir, Remdesivir, Tenofovir, Ribavirin, Galidesivir, 5c3, 5h1 and 7a1, show strong to moderate hydrogen bonding and hydrophobic interactions (π–π stacked, π–π T-shaped, π-sigma and π-alkyl). The stability provided from such interactions translate into greater antiviral activity or inhibitory effect of the ligands. Assessment of the average free energies of binding of the FDA approved drugs are highly comparable for conformers of a particular inhibitor, indicating similar modes of binding within the pockets.
Similar content being viewed by others
1 Introduction
Coronaviruses are a large group of RNA viruses that cause respiratory diseases and even hepatitis (Masters 2006; Spaan et al. 1988). They are widely distributed among mammals and birds (Masters 2006). There are many identified types of coronaviruses, which are about 25 species and are broadly classified into four groups—Alpha, Beta, Gamma and Delta coronaviruses. This classification is based on their genetic and serological relationship (Corman et al. 2018). There are now seven coronaviruses known to infect humans—HCoV-OC43, -229E, -NL63, -HKU1, SARS-CoV, MERS-CoV and now SARS-CoV-2 (Andersen et al. 2020; Corman et al. 2018). Its action is similar to SARS-CoV (Severe Acute Respiratory Syndrome—Coronavirus) and MERS-CoV (Middle East Respiratory Syndrome—Coronavirus) coronaviruses (Andersen et al. 2020). According to World Health Organization (WHO), most people infected with SARS-CoV-2, the virus that causes COVID-19, will experience mild to moderate respiratory illness and will intensify in older people and those with underlying medical problems such as cardiovascular disease, diabetes and cancer (WHO 2020).
Survival of SARS-CoV-2 outside the living system depends on the medium of exposure, contact time and temperature (van Doremalen et al. 2020). Studies with cryogenic electron microscope show that SARS-CoV-2 binds to the cell membrane of angiotensin-converting enzyme-2(ACE2) (Walls et al. 2020; Yang et al. 2013). This virus is enveloped RNA virus with a single, positive-strand RNA genome, which is reported to contain about 29,891–29,903 nucleotides (Walls et al. 2020). The genome carries two large replicase polyproteins that undergo proteolytic processing into a set of mature non-structural proteins (nsp) that function during viral replication, including RdRp, 3CLpro, Papain-like proteinase (PL2pro) and a superfamily 1-like helicase (HEL1) (Cheng et al. 2005).
Andersen et al. (2020) have identified key genomic features of SARS-CoV-2 that are adapted for entry into human host—the structure is optimized for binding to the human receptor cell known as angiotensin-converting enzyme 2(ACE2). Structurally, coronaviruses contain four main proteins in their genomes, namely S (spike protein), M (membrane protein), E (envelope protein), and N (nucleocapsid protein) and all have been identified in the SARS-CoV-2 proteome. The N protein is present in the nucleocapsid, M and E are the transmembrane proteins, while the S protein is the spike protein that is responsible for gaining entry into the host cell (Hasan & Hossain, 2020). Its Spike glycoprotein consists of two connected halves, S1–S2, which have to be separated by the furin enzyme on ACE2 cell of the human, by the insertion of 12 nucleotides (Walls et al. 2020).
SARS-CoV-2, therefore, selectively binds through S2 protein to the surface of ACE2 and from there been able to gain access to the human cell (Huang et al. 2006; Li et al. 2003; Prabakaran et al. 2004). MERS-CoV, SARS-CoV and SARS-CoV-2 are suggested to have originated from bats. However, while MERS-CoV have their reservoir in dromedary camels, SARS-CoV have their reservoir in bats. MERS-CoV, SARS-CoV and SARS-CoV-2 belong the same genus, the β-coronavirus. From this genus, the following SARS-CoV are reported to be endemic in humans: HCoV-OC43, HCoVHKU1, HCoV-NL63, and HCoV-229E (Chan and Chan 2013).
Viral polymerases are essential for viral genome replication and are targets for antiviral drug development (Cheng et al. 2005; Martin et al. 2017; Patil et al. 2019; Tomar et al. 2019). Since RdRp is an important protease that catalyzes the replication of RNA from RNA template, it is an attractive therapeutic target (Cheng et al. 2005). In HIV-1 RT, the inhibitors either bind to the hydrophobic pocket close to the polymerase or to the hydrophobic pocket on the surface of the thumb.
As scientists try to come up with vaccines or treatments for COVID-19, it becomes necessary to assess the binding interactions of some potential anti-viral agents against SARS-CoV-2 and SARS-CoV. One class of medicines that have held enormous therapeutic ability is the nucleotide-based (Flierl et al. 2015). They have often been modified to improve their stability and reduce degradation through, particularly, phosphorothioate backbone modification of nucleotide-based drugs (Erion et al. 2006; Flierl et al. 2015).
Some antiviral medications approved by the Food and Drug Administration (FDA) have been tested, while others have been suggested to inhibit SARS-CoV-2 (Gao et al. 2020a, b; Touret and de Lamballerie 2020). Ju et al. (2020a, b) note that coronaviruses replicate in the same way as hepatitis C virus and describe novel strategies to design and synthesize nucleotide analogues, viral polymerase inhibitors. These nucleotides show potent polymerase inhibitory effect when the active forms of Sofosbuvir and Alovudine are incorporated to terminate further nucleotide extension by RdRp in the polymerase reaction (Ju et al. 2020a, b). Also, Remdesivir, adenosine nucleotide analogue, is reported to act as a broad-spectrum antiviral drug. However, its mechanism of action is not well known (Ju et al. 2020a, b; Wang et al. 2020a, b).
This in silico study, therefore, assesses the binding affinity of nucleotide analogues designed by Ju et al. (2020a, b) and compares their binding energies and interactions with selected antiviral drugs approved by the FDA. Other drug molecules considered in this work are Quinine (effective against Plasmodium falciparum, the parasite that causes malaria), Thymoquinone, Ivermectin, antivirals drugs such as Oseltamivir, Sofosbuvir, Tenofovir and Peramivir (Cao et al. 2014). In addition, other compounds that have also been studied include; hydroxychloroquine, ribavirin, remdesivir, favipiravir and galidesivir. These have been selected, because recent reports that show their activity against viruses and possibly SARS-CoV-2 and SARS-CoV (Colson et al. 2020; Li and De Clercq 2020; Wang et al. 2020a, b).
2 Materials and methods
2.1 Preparation of RdRp templates
The “FASTA” (Pearson 1994) query sequences of SARS-CoV-2 and SARS-CoV nucleotide genes were obtained from the NCBI (National Centre for Biotechnology Information) repository via https://www.ncbi.nlm.nih.gov. Their reference sequence numbers are NC_045512.2 and NC_004718.3 for SARS-CoV-2 and SARS-CoV nucleotides, respectively. The electronic crystallographic structures of SARS-HCoV RdRp (PDB Code: 6NUR, Chain A, resolution 3.1 Å) and SARS-CoV-2RdRp (PDB Code: 6M71, Chain A, resolution 2.9 Å) were used as templates to develop the homology models for SARS-CoV-2 and SARS-CoV RdRp, respectively. With the query sequence and templates in hand, homology models were developed for SARS-CoV and SARS-CoV-2 via the SWISS-MODEL webserver (Waterhouse et al. 2018). Furthermore, the integrity of the obtained models was verified by structure validation on SAVES v5.0 (Beg et al. 2018; Lüthy et al. 1992) server of the University of California, Los Angeles.
2.2 Preparation of 3CLpro templates
In another molecular docking experiment, to study the interaction of the inhibitors with chymotrypsin-like cysteine protease (3CLpro), crystal structures of SARS-CoV-2 (PDB code: 6LU7, resolution 2.16 Å) and SARS-CoV (PDB code: 1UJ1, resolution 1.9 Å) were used. All the PDB files used in this work were downloaded in.pdb format from the Protein Data Bank at http://www.rcsb.org.
2.3 Molecular docking protocol
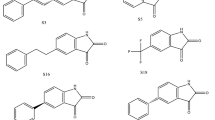
Molecular docking was carried out using Iterated Local Search global optimizer implemented in AutoDock Vina (Trott and Olson 2010) with the help of a graphical user interface program, PyRx (Dallakyan and Olson 2015). Free water molecules were deleted from the crystallographic structures of 3CLpro proteins, while the RdRp model structures of both SARS-CoV and SARS-CoV-2 were used as obtained from SWISS-MODEL webserver. All the protein structures were examined for gabs and hydrogen added before docking simulation. Throughout the docking experiment, the protein structures were kept rigid, while the torsions or degrees of freedom for the ligands were allowed full rotations. Conformational space for docking simulation was sampled following the literature reports of known active sites for 1UJ1 and 6LU7 3CLpro proteins (Jin et al. 2020; Yang et al. 2003). The ligand-binding sites for SARS-CoV and SARS-CoV-2 RdRps are shown in Fig. 3, similar to the reports of (Gao et al. 2020a, b; Kirchdoerfer and Ward 2019). A total of 173 inhibitors are considered in this molecular docking study and their structural representations are given in Figs. 1 and 2. The grid dimensions that were used are contained in Supplementary Information (SI)1. The visualization tools used in this work are UCSF Chimera (Pettersen et al. 2004) and Discovery Studio 2017 R2.

Non FDA approved nucleotide analogues. 1–4. a R1 = methyl (CH3), b R1 = ethyl (CH2CH3), c R1 = allyl (CH2CH = CH2), d R1 = propyl (CH2CH2CH3),), e R1 = methoxymethyl (CH2OCH3), f R1 = propargy (CH2CCH), g R1 = methylthiomethyl (CH2SCH3), h R1 = azidomethyl (CH2–N3). 5–8. a1 R2 = H, R1 = a; a2 R2 = OH, R1 = a; a3 R2 = F, R1 = a; a4 R2 = OCH3, R1 = a. b1 R2 = H, R1 = b; b2 R2 = OH, R1 = b; b3 R2 = F, R1 = b; b4 R2 = OCH3, R1 = b. c1 R2 = H, R1 = c; c2 R2 = OH, R1 = c; c3 R2 = F, R1 = c; c4 R2 = OCH3, R1 = c. d1 R2 = H, R1 = d; d2 R2 = OH, R1 = d; d3 R2 = F, R1 = d; d4 R2 = OCH3, R1 = d. e1 R2 = H, R1 = e; e2 R2 = OH, R1 = e; e3 R2 = F, R1 = e; e4 R2 = OCH3, R1 = e. f1 R2 = H, R1 = f; f2 R2 = OH, R1 = f; f3 R2 = F, R1 = f; f4 R2 = OCH3, R1 = f. g1 R2 = H, R1 = g; g2 R2 = OH, R1 = g; g3 R2 = F, R1 = g; g4 R2 = OCH3, R1 = g. h1 R2 = H, R1 = h; h2 R2 = OH, R1 = h; h3 R2 = F, R1 = h; h4 R2 = OCH3, R1 = h (Ju et al. 2020a, ba)

2.4 Preparation of inhibitor structures
Full geometry optimizations of the molecular structures of the inhibitors (ligands), without symmetry constraints were performed. Stationary points on the potential energy surface were characterized, ascertained to have no imaginary frequency. All calculation Density Functional Theory (DFT) calculations were performed using Gaussian 16 suite of programs by means of resources provided by SEAGrid (Pamidighantam et al. 2016) facilities. DFT calculations were performed at the B3LYP (Lee et al. 1988)/6-31G(d) level of theory.
3 Results and discussion
3.1 Structure validation
The model structures obtained from the homology modeling using SWISS-MODEL (Biasini et al. 2014) were subjected to a series of evaluations and validation using MolProbity (Williams et al. 2018) and the different modules of SAVES v5.0; VERIFY, ERRAT and PROCHECK. For SARS-CoV-2 and SARS-CoV, 97.50% and 96.05% of the respective Ramachandran plots are in favoured regions and their corresponding MolProbity values are 1.03 and 0.66. The Ramachandran plots are presented in Fig. 7 and 8 of supplementary information (SI2). VERIFY was also employed in validation; for SARS-CoV-2 (89.66% of the residues have averaged 3D-1D score ≥ 0.2) and for SARS-CoV (89.20% of the residues have averaged 3D-1D score ≥ 0.2). ERRAT gave an overall quality factor for SARS-CoV-2 and SARS-CoV as 95.905 and 95.594, respectively. The sequence alignment of SARS-CoV 3CLpro (PDB Code: 6LU7) and SARS-CoV-2 3CLpro (PBD Code: 1UJ1) gave percent identity of 93.79% (Fig. 9a of SI2) and the sequence alignment of SARS-CoV RdRp with SARS-CoV-2 RdRp shows 95.89% identity, in Fig. 9b of SI2.
3.2 Aligning the homology model
The model structures and their respective templates were aligned, Fig. 6 of the SI2, with SARS-CoV-2 and SARS-CoV giving RMSDs of 0.144 and 0.069 Å. Similarly, the aligned structures displayed in Fig. 3 indicate that SARS-CoV-2 and SARS-CoV RdRp are closely related, having RMSD of 0.501 Å, with 762 conserved residues, with 95.89% identity. These structures also show features that are conserved due to similar protein conformations, particularly in the four critical regions—Thumb, Finger, N-terminal and Palm. The active sites of the SARS-CoV and SARS-CoV-2 RdRp proteins are conserved and are formed by conserved polymerase motifs located around the palm domain. However, they are structurally different around the N-terminal, as indicated by the “less conserved N-terminal” in Fig. 3.

Aligned model structures of SARS-CoV-2 (red) with SARS-CoV (blue) RdRp showing the four catalytic domains; palm, thumb, finger and N-terminal (color figure online)
3.3 Analysis of docking results
COACH meta-server (Yang et al. 2013) identifies the consensus binding residues for SARS-CoV-2 RdRp model to include the following VAL477, PHE482, TYR485, ARG571, ARG572, GLN575, ARG642, LYS643, HIS644, ASN697, CYS699, GLY714. The residues that are within 5.0 Å of the active site of the template, 6NUR, are CYS647, CYS489, ILE307, LEU302, CYS301, THR293 ASN200. During the docking experiment, the entire active site conformational space was sampled to examine the kind of interactions that might occur around the active site and other pockets. Similarly, COACH also identifies the consensus residues of ligand binding to include; LYS426, ILE466, LEU470, TYR515, MET519, ASP523, ASP525, ALA526, VAL535, ILE539, ASN543, ALA547, SER561, VAL587, ILE589, LYS593, PHE594, HIS650, PHE652, TYR653, ARG654, LEU655, 657, 658, CYS659, GLN661, GLU729, CYS730, LEU731, and SER768.
3.4 Gibb’s free energies of binding
Generally, the free energies of binding are calculated as a sum of four energy terms, namely, intermolecular free energy (dominated by enthalpic contribution from steric and electrostatic interaction upon complex formation), energy of solvation, free energy associated with the motion of ligand, protein and ligand–protein complex and free energy due to conformational changes (Raha & Merz, 2005). However, AutoDock Vina scoring function was inspired by the scoring function implemented in X-score and is given in Eq. 1:
where \(\Delta G_{{{\text{vdw}}}} \) takes care of free energy contribution due to van der Waals interaction between the ligands and the proteins; \(\Delta G_{{{\text{H}} - {\text{bond}}}}\) accounts for the hydrogen bonding between the ligand and the protein; \(\Delta G_{{{\text{deformation}}}}\) accounts for the free energy of deformation during docking; \(\Delta G_{{{\text{hydrophobic}}}}\) accounts for the hydrophobic interaction during simulation and \(\Delta G_{0}\) is the regression constant that includes translational and rotational entropy loss in binding (Trott and Olson 2010; Wang et al. 2002).
During each docking process, nine conformational modes were generated and the best conformers, those with the lowest free energies of binding, are presented in Table 1. From the results, it is easy to see that the free energies of binding are strongly negative which indicates that the inhibitors bind tightly to the active residues of proteins used in the study. The difference in the standard deviations of the nine conformational modes of each inhibitor is not much, indicating that the binding affinity of each conformer does not differ greatly from others. Similarly, the mean free energies of binding are highly comparable for conformers of a particular inhibitor, as can be seen in SI1.
Generally, 3CLpro of SARS-CoV-2 show higher binding energies compared to the other protein structures with the same inhibitors. We use here the result of Remdesivir as positive control since it has been chosen by the WHO for clinical trials to develop medication for SARS-CoV-2. For 3CLpro protein of SARS-CoV-2, the binding energies for the ligands are higher than values for Remdesivir and Ivermectin, with values − 7.4 and − 8.4 kcal/mol, respectively. Similar comparison is done for its SARS-CoV counterpart. Except for Sofosbuvir and Ivermectin, with binding energy of − 8.2 and − 8.1 kcal/mol, respectively, the rest of the inhibitors give binding energies higher than Remdesivir. To investigate the possible reasons why ivermectin bind well than Remdesivir, we examined the kinds of interactions that assist in protein–ligand complexes. SARS-CoV-2-Ivermectin complex shows mainly carbon-hydrogen bonding at 3.5 Å while in the protein-Remdesivir complex, 6 hydrogen bonds (HB’s) are formed at a range of 2.2–3.48 Å and 3 hydrophobic interactions. Since hydrophobic interactions are relatively stronger than other weak intermolecular forces, this translates into better binding for Ivermectin (− 8.4 kcal/mol) compared to Remedesivir (between − 7.9 and − 7.0 kcal/mol) and other FDA-approved nucleotide prodrugs when interacted with 3CLpro and RdRp of SARS-CoV and RdRp of SARS-CoV-2. However, Remdesivir binds stronger with SARS-CoV RdRp protein, with an affinity of − 8.7 kcal/mol than Ivermectin with a binding affinity of − 8.4 kcal/mol. Analysis of the bound complexes shows that electrostatic interaction plays a major role in the binding of Remdesivir with ASP218 and LYS50 residues. Asp218, known as the nidovirus conserved residue, is reported to play a critical role in the virus activity (Kirchdoerfer and Ward 2019). The number of HB’s and other hydrophobic interactions (such as pi-alkyl) increase in both SARS-CoV RdRp—Remdesivir and SARS-CoV RdRp—Ivermectin complexes. Binding energy of Vitamin C is relatively higher when compared to other inhibitors. This is expected as Vitamin C is a known immune system booster against virus attack and not an antiviral drug. In general, the binding energies of the RdRp homologues are highly comparable, with SARS-CoV showing slightly lower values, with few exceptions like Ivermectin and Galidesivir. However, Quinine with value of − 7.3 kcal/mol binds to SARS-CoV-2 better than Remdesivir, so also Peramivir, − 7.2 kcal/mol, Sofosbuvir, – 7.7 kcal/mol and Ivermectin, − 8.8 kcal/mol. It is interesting to note that our result is in excellent agreement with a recent report (Caly et al. 2020) that Ivermectin, an FDA-approved anti-parasitic, previously shown to have broad-spectrum anti-viral activity in vitro, is an inhibitor of the causative virus (SARS-CoV-2).With a single addition to Vero-h SLAM cells 2 h post-infection with SARS-CoV-2 is able to effect ∼ 5000-fold reduction in viral RNA at 48 h. Ivermectin is, therefore, currently in the frontline as one of the drugs being considered as possible treatment for COVID-19 infection (Chaccour et al. 2020).
Antiviral drugs that are polymerase inhibitors such as Sofosbuvir, Remdesivir, Tenofovir, Ribavirin and Galidesivir are purine nucleotide prodrugs that are considered in this investigation. Syntheses of their analogues used in this work, Fig. 1, have been described by Ju et al. (2020a, b). The analogue show very promising inhibitory potentials when considering their binding energies. Generally, the binding energies of the inhibitors including the nucleotide analogues, range between − 5.4 and − 8.4 kcal/mol for SARS-CoV-2 RdRp, − 4.9 and − 8.7 kcal/mol for SARS-CoV RdRp, − 5.5 and − 8.6 kcal/mol for SARS-CoV 3CLpro and − 5.1 and − 8.4 kcal/mol for SARS-CoV-2 3CLpro. However, Ivermectin recorded the highest binding affinity, with a ΔG of − 8.8 kcal/mol, while Thymoquinone gave one of the lowest binding affinities, with a value of − 5.4 kcal/mol.
3.5 Chemistry of binding of the HCoV structures with inhibitors
Several kinds of chemical interactions mediate the differences in the calculated free energies of binding between pocket amino acid residues and the molecular inhibitors that are used in this investigation. Some of the interactions include electrostatic interactions (HB), hydrophobic and halogen bonding. HB in this 3CLpro SARS-CoV-2-ligand-binding interaction is mainly due to the presence of amide groups in the organic backbone of the protein, which act as sites for H-bond donor and acceptor. Pertinent to note is that the various non-covalent interactions that occur, apart from HB, within the protein–ligand complex in our study are weak but play a major role in determining the stability of the system. Electrostatic interactions that are observed are generally charge (attraction and repulsion) and π-cation. The hydrophobic interactions that are observed are π-sigma, π-πstacking, π–π T-shaped and π-alkyl.
3.5.1 3CLpro of SARS-CoV-2—ligand-binding interactions
Non-covalent interactions are the major chemical interaction observed, particularly HB between the ligands and protein residues. HB is important in protein–ligand interaction, because it leads to the structure stability (Panigrahi 2008). The classification scheme for the H-bonding interactions described in this work is Jeffrey’s (Jeffrey 1997). They are divided into three, namely, strong and mostly covalent (2.2–2.5 Å), moderate and electrostatic (2.5–3.2 Å) and weak and electrostatic (3.2–4.0). These hydrogen bonds, therefore, are formed between an electronegative atom and a hydrogen atom bonded to a second electronegative atom (Henderleiter et al. 2001). In protein, the H-bonding are mainly within the moderate region and shifts to either side due to the kind of moiety that is present in the environment of the protein.
The nature of the chemical interactions of the ligands (Figs. 1 and 2) with the proteins of HCoVs that are investigated here are now discussed. Sheet 1 of SI1 shows that apart from the HB interactions that are generally observed, other interactions further contribute to a decrease in the energy of binding of the complex. This can be observed in Remdesivir and Sofosbuvir, among the FDA approved antiviral drugs and also in the non-FDA approved nucleotide analogues (Ju et al. 2020a, b), Fig. 1, (5–8). They are here described as non-FDA approved drugs since they are recently synthesized and have not even undergone clinical trials. 5c3, 5h1 and 7a1 show strong HB which occur within a range of 2.20–3.02 Å, and give a low binding energy, ΔG = – 8.4 kcal/mol. While 5c3 and 7a1 are π-stacked with HIS41 (active residue) over a distance of 4.33 and 4.41 Å, respectively, 5h1 rather interacts with HIS41 via a non-conventional C-H bonding within 3.5 Å. The spatial distance of the benzene rings of both 5c3 and 7a1 from HIS41′s is large enough to reduce the electrostatic interactions between the π-electrons of the interacting benzene rings (Fu et al. 2016). As expected of such interaction, the bond type is weak and acidic behavior will dominate (Spiwok 2017). π-sulphur is another non-covalent interaction that occurs where the π-electron cloud of MET165 interacts with the lone pair of electron cloud of the sulphur atom. Remdesivir, which is chosen as reference here, show π-orbital interaction with MET165 (active residue) at 5.74 Å. Pertinent is the π–π stacked interaction between the electron cloud of the benzene ring in HIS41 and the central aromatic ring of Remdesivir at a distance of 4.00 Å. This is 0.34 Å higher than 3.66 Å, for which an equivalent value would be enough to cause 7.5 eV rise in energy of the system (Fu et al. 2016). This buttresses the fact that π-π stacking at large distances stabilizes the protein–ligand complex by reducing the repulsive electrostatic interaction. Strong HB interactions that are observed lie between 2.12 and 2.87 Å. In addition to the HB with GLY143, GLN189 and 142, Remdesivir made hydrophobic contacts with HIS41, LEU27 and MET49. The weak attractive charge interaction is due to P-atom of the nucleoside phosphoramidite group and ASP289 (OD2, 4.3 Å), an acidic residue, which results in minimal stability at site of interaction (Bajji and Davis 2000) and the conventional HB are due to ARG131 (HH2, 2.80 Å) as donors to the oxo-benzenoid group of the Sofosbuvir.
Quinine and Galidesivir, with the same binding score (− 6.7 kcal/mol) are dominated mainly by HB but Galidesivir shows π-π T-shaped hydrophobic interaction due to the π-electron cloud of its pyrryl ring with HIS41 in the active site, as shown in Fig. 4. This contributes to the extra stability of the protein–ligand complex (Martinez and Iverson 2012).

Docked Galidesivir molecule showing π-π T-shaped interaction with HIS41active residue of SARS-CoV-2 3CLpro
Oseltamivir interacts with the residues of the pocket, howbeit, through HB with LYS137 (HZ2, 2.11 Å; HZ23, 2.63 Å) and LEU287 (O, 2.35 Å). Even though conventional HB interaction was not observed, most of the interactions observed are non-covalent, electrostatic except non-conventional C-H (CG2, 3.91 Å) bonding with ILE106.
3.5.1.1 CLpro of SARS-CoV-ligand-binding interactions
Generally, AutoDock Vina binding scores for 3CLpro of SARS-CoV are slightly better than 3CLpro of SARS-CoV-2. From sheet 2 of SI1, the free energy of binding ranges from − 5.5 to − 8.6 kcal/mol. The structures of HCoV 3CLpros have been described clearly elsewhere, (Chen et al. 2020; Cheng et al. 2005) hence, only their salient features would be highlighted here. 3CLpro structures of SARS-CoV and SARS-CoV-2 are dimers with three domains, I, II and III as shown in Fig. 5, for HCoV-2–Remdesivir complex. The catalytic mechanism does not follow the triad seen in other chymotrypsin protease, comprising SER-HIS-ASP (Shi et al. 2008; Solowiej et al. 2008), rather a catalytic dyad comprising CYS145 and HIS41 (Khan et al. 2020; Shi et al. 2008), which lie between domains I and II. It is very intriguing to discover that the FDA-approved antiviral prodrugs considered in this work do not make simultaneous contact with the dyad. Rather, most of the non-clinically tried nucleotide analogues made consistent, simultaneous contacts through the HE2 and SG subdomains of HIS41 and CYS145, respectively.

Structure of SARS-CoV-2 3CLpro, showing the three catalytic domains. The insert shows Remdesivir docked in the pocket between domains I and II, in contact with HIS41 and CYS145 also within the pocket
Assessment of the inhibitor with the best binding affinity, 2a with ΔG = − 8.6 kcal/mol, shows HB interactions lying between 2.01 and 3.51 Å and hydrophobic interaction with the residues. Its major HB interactions are with the following residues GLN110 (HE21, 3.00 Å; OE1, 2.01 Å), ARG105, PRO293, THR111, ASN151 and ASP295 and the major hydrophobic interactions are π-sigma (C–H, 3.29 Å) with LEU202, π-orbital interaction occur over long distances; PHE294 (5.24 Å), ILE249 (5.47 Å) and VAL104 (5.24 Å). These interactions provide extra-stability to the protein–ligand complex, resulting in a low ΔG. 3a with ΔG = -– 8.3 kcal/mol shows similar interactions as can be seen in SI1.
Interesting to note is that hydrophobic interaction contributes to low binding energy of Sofosbuvir against 3CLpro of SARS-CoV. with similar observation for Remdesivir (– 7.9 kcal/mol) and
Generally, the FDA-approved antiviral drugs bind with relatively higher ΔG values and mainly through HB but the non-FDA approved nucleotide analogues show other forms of non-covalent interactions and this contributes to the relative better binding energies. Details of all the interactions observed for both 3CLpro nCov’s are shown in sheets 1 and 2 of SI1.
Even though conventional H-bonding interaction was not observed in Hydroxychloroquine (HQ), most of the interactions observed are electrostatic except weak C-H (CG2, 3.91 Å) bonding interaction between IL4106 and -OH group of the ligand. The interaction of the HQ is also mediated by hydrophobic contacts with ILE106 and VAL104 through π-alkyl mode of interaction (Fantini et al. 2020). These hydrophobic interactions underscore the low binding affinity for 3CLpro of SARS-CoV-2.
3.5.2 SARS-CoV-2 RdRP and SARS-CoV RdRp
A repurposing attempt has been made in this research, since it is of advantage in formulating and developing drugs from the clinically approved types so as to be able to improve on their functionality. In that light, the Remdesivir and other nucleotidenon preclinically tried drug candidates (Ju et al. 2020a, b) have also been tested for their efficacy in reversing the RNA polymerase action of nCoV’s.
With the set of compounds under investigation, the binding energy ranges − 5.4 to − 8.8 kcal/mol for SARS-Cov-2 RdRp protein and − 4.9 to − 8.7 kcal/mol for SARS-CoV RdRp. Since the range of binding energy is almost the same, it shows that the action of these antiviral polymerase drug developed for SAR-CoV-2 RdRp would be the same for SARS-CoV RdRp. Hence, one could be used for the treatment of the other. However, it is worth examining the modes of interaction of non-clinically aproved drugs in this in silico study with the clinically approved.
Hydrogen bonds alone mediate the binding of Ribavirin to the acidic residues, ASP762 (H12, 2.4 Å), and neutral groups [TYR621 (HN, 2.34 Å) and TRP802 (HE1, 2.39 Å)], while there is π-cation attraction observed in Remdesivir, Sofosbuvir, Quinine, Paracetamol and Favipiravir for ARG626. These interactions help in binding to the active sites of the RdRp of SARS-CoV-2 and SARS-CoV, and are stabilised by hydrophobic contacts.Such hydrophobic interaction of immense effect is π-π stacking with the residue, TYR730 (\(\sim 4.1 {\AA}\)), and ligand π-electron clouds of the aromatic systems.
The best binding energy values for non-approved nucleotide drug candidates (SI1, sheet 3), have been clustered into four, for SARS-CoV-2 RdRp thus; -8.2 kcal/mol (7f3, 7b3 and 7e3), − 8.1 kcal/mol (7a3, 7b1, 1c and 3a), − 7.9 kcal/mol (7c1, 5h1, 6f2, 2b, 3h, 1h, 5a3 and 6a3), and − 7.8 kcal/mol 1d, 4c,7c3, 6f1, 5b1, 5c1, 7g1, 6a4, 7f2, 7d2 and 7e2). The dominant interactions give rise to non-covalent stabilising effects, which also result in high binding affinity for RdRp proteins. The hydrophobic interacting residues are ARG351, VAL677, TYR131, which are mainly due to π-orbital–π-orbital, alky-alky and π-orbital–alkyl effects. However, similar observations are made for SARS-CoV RdRp.
4 Conclusion
Homology modelling has been reported for RdRp structures of SARS-CoV and SARS-CoV-2. The structural alignment of both HCoVRdRp’s gave an RMSD of 0.461 Å with 761 conserved residues. Structural alignment of the SARS-CoV and SARS-CoV-2 RdRps only differ at the N-terminal, close to the palm domain and are highly conserved in the other domains. Since the free energies of binding of the various inhibitors to the RdRps are highly comparable, any medication for one can be used for the treatment of the other. However, assessment of the chemical interactions with RdRps and 3CLpro’s reveals stronger hydrogen bonding and non-covalent effects in RdRp’s, suggesting that the nucleotide prodrugs are more effective against their target than in 3CLpro’s. Since the HCoV protease cleave the precursor forms of the helicase before initiating the formation of replication–transcription complex, the energies of binding to the different structures of HCoV strains indicate that the FDA-approved antiviral drugs—Remdesivir and Sofosbuvir—better yet their analogues used in this work, can stop or reverse the polymerase function of these viruses and hence serve as potential drug candidates.
References
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 89(1):44–48. https://doi.org/10.1038/s41591-020-0820-9
Bajji AC, Davis DR (2000) Synthesis and biophysical characterization of tRNALys,3 anticodon stem-loop RNAs containing the mcm5s2U nucleoside. Org Lett 2(24):3865–3868. https://doi.org/10.1021/ol006605h
Beg MA, Shivangi TSC, Meena LS (2018) Structural Prediction and mutational analysis of Rv3906c gene of Mycobacterium tuberculosis H37Rv to determine its essentiality in survival. Adv Bioinform. https://doi.org/10.1155/2018/6152014
Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y (2012) Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol 42(2):145–153
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42(W1):W252–W258. https://doi.org/10.1093/nar/gku340
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM (2020) The FDA-approved Drug Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res. https://doi.org/10.1016/j.antiviral.2020.104787
Cao R-Y, Xiao J-H, Cao B, Li S, Kumaki Y, Zhong W (2014) Inhibition of novel reassortant avian influenza H7N9 virus infection in vitro with three antiviral drugs, oseltamivir, peramivir and favipiravir. Antiviral Chem Chemother 23(6):237–240. https://doi.org/10.3851/IMP2672
Chaccour C, Hammann F, Ramón-García S, Rabinovich NR (2020) Ivermectin and Novel Coronavirus Disease (COVID-19): Keeping Rigor in Times of Urgency. Am J Trop Med Hygiene. https://doi.org/10.4269/ajtmh.20-0271
Chan PKS, Chan MCW (2013) Tracing the SARS-coronavirus. J Thorac Dis 5 Suppl 2(Suppl 2):S118–S121. https://doi.org/10.3978/j.issn.2072-1439.2013.06.19
Chen YW, Yiu C-PB, Wong K-Y (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL (pro)) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 9:129. https://doi.org/10.12688/f1000research.22457.1
Cheng A, Zhang W, Xie Y, Jiang W, Arnold E, Sarafianos SG, Ding J (2005) Expression, purification, and characterization of SARS coronavirus RNA polymerase. Virology 335(2):165–176. https://doi.org/10.1016/j.virol.2005.02.017
Colson P, Rolain JM, Lagier JC, Brouqui P, Raoult D (2020) Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2020.105932
Corman VM, Muth D, Niemeyer D, Drosten C (2018) Hosts and sources of endemic human coronaviruses. In: Advances in virus research, vol. 100. Elsevier Inc., pp 163–188. https://doi.org/10.1016/bs.aivir.2018.01.001
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx BT—chemical biology: methods and protocols. In: Hempel JE, Williams CH, Hong CC (eds) Chemical biology. Springer New York, pp 243–250. https://doi.org/10.1007/978-1-4939-2269-7_19
Erion MD, Bullough DA, Lin C-C, Hong Z (2006). HepDirect prodrugs for targeting nucleotide-based antiviral drugs to the liver. Curr Opin Investig Drugs (London, England : 2000) 7(2): 109–117. http://europepmc.org/abstract/MED/16499280. Accessed 23 Apr 2020
Fantini J, Di Scala C, Chahinian H, Yahi N (2020) Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2020.105960
Flierl U, Nero TL, Lim B, Arthur JF, Yao Y, Jung SM, Gitz E, Pollitt AY, Zaldivia MTK, Jandrot-Perrus M, Schäfer A, Nieswandt B, Andrews RK, Parker MW, Gardiner EE, Peter K (2015) Phosphorothioate backbone modifications of nucleotide-based drugs are potent platelet activators. J Exp Med 212(2):129–137. https://doi.org/10.1084/jem.20140391
Fox RI, Dixon R, Guarrasi V, Krubel S (1996) Treatment of primary Sjögren's syndrome with hydroxychloroquine: a retrospective, open-label study. Lupus 5(1_suppl):31–36
Fu X-X, Li J-F, Zhang R-Q (2016) Strong orbital Interaction in pi-pi Stacking System. In: arXiv e-prints arXiv:1601.01150. https://ui.adsabs.harvard.edu/abs/2016arXiv160101150F. Accessed 23 Apr 2020
Gao J, Tian Z, Yang X (2020a) Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends 14(1):72–73. https://doi.org/10.5582/bst.2020.01047
Gao Y, Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Wang Q, Lou Z, Rao Z (2020b) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368(6492):779–782
Hasan S, Hossain MM (2020) Analysis of COVID-19 M protein for possible clues regarding virion stability , longevity and spreading. https://doi.org/10.31219/osf.io/e7jkc
Henderleiter J, Smart R, Anderson J, Elian O (2001) How do organic chemistry students understand and apply hydrogen bonding? J Chem Educ 78(8):1126. https://doi.org/10.1021/ed078p1126
Huang IC, Bosch BJ, Li F, Li W, Kyoung HL, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M, Rottier PJM, Choe H (2006) SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281(6):3198–3203. https://doi.org/10.1074/jbc.M508381200
Jeffrey GA (1997) An introduction to hydrogen bonding (12th ed.). Oxford University Press. https://books.google.fr/books?id=ZRAFifo37QsC. Accessed 23 Apr 2020
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang H (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
Ju J, Kumar S, Li X, Jockusch S, Russo JJ (2020a) Nucleotide analogues as inhibitors of viral polymerases. BioRxiv. https://doi.org/10.1101/2020.01.30.927574
Ju J, Li X, Kumar S, Jockusch S, Chien M, Tao C, Morozova I, Kalachikov S, Kirchdoerfer RN, Russo JJ (2020b) Nucleotide Analogues as Inhibitors of SARS-CoV Polymerase. BioRxiv. https://doi.org/10.1101/2020.03.12.989186
Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z (2020) Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn 9(1):1–10. https://doi.org/10.1080/07391102.2020.1751298
Kirchdoerfer RN, Ward AB (2019) Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 10(1):1–9. https://doi.org/10.1038/s41467-019-10280-3
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785–789. https://doi.org/10.1103/PhysRevB.37.785
Li G, De Clercq E (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19(3):149–150. https://doi.org/10.1038/d41573-020-00016-0
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454. https://doi.org/10.1038/nature02145
Lüthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356(6364):83–85. https://doi.org/10.1038/356083a0
Martin B, Canard B, Decroly E (2017) Filovirus proteins for antiviral drug discovery: Structure/function bases of the replication cycle. Antivir Res 141:48–61. https://doi.org/10.1016/j.antiviral.2017.02.004
Martinez CR, Iverson BL (2012) Rethinking the term “pi-stacking.” Chem Sci 3(7):2191–2201. https://doi.org/10.1039/C2SC20045G
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 65(06):193–292. https://doi.org/10.1016/S0065-3527(06)66005-3
Pamidighantam S, Nakandala S, Abeysinghe E, Wimalasena C, Yodage SR, Marru S, Pierce M (2016) Community science exemplars in SEAGrid science gateway: apache airavata based implementation of advanced infrastructure. Proc Comput Sci 80:1927–1939. https://doi.org/10.1016/j.procs.2016.05.535
Panigrahi SK (2008) Strong and weak hydrogen bonds in protein-ligand complexes of kinases: a comparative study. Amino Acids 34(4):617–633. https://doi.org/10.1007/s00726-007-0015-4
Patil VM, Balasubramanian K, Masand N (2019) Chapter 14—dengue virus polymerase: a crucial target for antiviral drug discovery. In: Gupta SPBT-VP (ed) Viral polymerases. Academic Press, pp 387–428. https://doi.org/10.1016/B978-0-12-815422-9.00014-0
Pearson WR (1994) Using the FASTA program to search protein and DNA sequence databases. In: Griffin AM, Griffin HG (eds) Computer analysis of sequence data. Humana Press, pp 307–331. https://doi.org/10.1385/0-89603-246-9:307
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605
Prabakaran P, Xiao X, Dimitrov DS (2004) A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem Biophys Res Commun 314(1):235–241. https://doi.org/10.1016/j.bbrc.2003.12.081
Raha K, Merz KMBT-ARC C (2005) Chapter 9 Calculating binding free energy in protein–ligand interaction. In: Annual reports in computational chemistry, vol. 1. Elsevier, pp. 113–130. https://doi.org/10.1016/S1574-1400(05)01009-1
Shi J, Sivaraman J, Song J (2008) Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J Virol 82(9):4620LP – 4629. https://doi.org/10.1128/JVI.02680-07
Solowiej J, Thomson JA, Ryan K, Luo C, He M, Lou J, Murray BW (2008) Steady-State and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro Cysteine protease: development of an ion-pair model for catalysis. Biochemistry 47(8):2617–2630. https://doi.org/10.1021/bi702107v
Spaan W, Cavanagh D, Horzinek MC (1988) Coronaviruses: structure and genome expression. J Gen Virol 69(12):2939–2952. https://doi.org/10.1099/0022-1317-69-12-2939
Spiwok V (2017) CH/π Interactions in Carbohydrate Recognition. Molecules. https://doi.org/10.3390/molecules22071038
Tomar S, Mudgal R, Pareek A (2019) Chapter 3—RNA-dependent RNA polymerase of alphaviruses: a potential target for the design of drugs against alphaviruses. In: Gupta SPBT-VP (ed) Viral polymerases. Academic Press, pp 69–94. https://doi.org/10.1016/B978-0-12-815422-9.00003-6
Touret F, de Lamballerie X (2020) Of chloroquine and COVID-19. Antivir Res 177:104762. https://doi.org/10.1016/j.antiviral.2020.104762
Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
van Doremalen N, Bushmaker T, Morris DH, Holbrook MG, Gamble A, Williamson BN, Tamin A, Harcourt JL, Thornburg NJ, Gerber SI, Lloyd-Smith JO, de Wit E, Munster VJ (2020) Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med 382(16):1564–1567. https://doi.org/10.1056/NEJMc2004973
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 180:1–12. https://doi.org/10.1016/j.cell.2020.02.058
Wang R, Lai L, Wang S (2002) Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des 16(1):11–26. https://doi.org/10.1023/A:1016357811882
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G (2020a) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30(3):269–271. https://doi.org/10.1038/s41422-020-0282-0
Wang Y, Wang Y, Chen Y, Qin Q (2020b) Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicate special control measures. J Med Virol. https://doi.org/10.1002/jmv.25748
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(1):296-W303. https://doi.org/10.1093/nar/gky427
WHO (2020) Coronavirus disease (COVID-2019) situation reports 318. https://doi.org/10.1093/nar/gkz966
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB III, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC (2018) MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27(1):293–315. https://doi.org/10.1002/pro.3330
Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 100(23):13190–13195
Yang J, Roy A, Zhang Y (2013) Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29(20):2588–2595. https://doi.org/10.1093/bioinformatics/btt447
Acknowledgements
We thank SEAGrid for the provision of computing facilities and the University of Lagos, Nigeria for providing support to the Physical Organic Chemistry research group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Udofia, I.A., Gbayo, K.O., Oloba-Whenu, O.A. et al. In silico studies of selected multi-drug targeting against 3CLpro and nsp12 RNA-dependent RNA-polymerase proteins of SARS-CoV-2 and SARS-CoV. Netw Model Anal Health Inform Bioinforma 10, 22 (2021). https://doi.org/10.1007/s13721-021-00299-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-021-00299-2