
Abstract
Four undescribed pyrethrins C-F (1–4) as well as four known pyrethrins (5–8) were isolated from seeds of Pyrethrum cinerariifolium Trev. The structures of compounds 1–4 were elucidated by UV, HRESIMS, and NMR (1H and 13C NMR, 1H-1H COSY, HSQC, HMBC and ROESY), among which the stereostructure of compound 4 was determined by calculated ECD. Furthermore, compounds 1–4 were evaluated for their aphidicidal activities. The insecticidal assay results showed that 1–4 exhibited moderate aphidicidal activities at the concentration of 0.1 mg/mL with the 24 h mortality rates ranging from 10.58 to 52.98%. Among them, pyrethrin D (2) showed the highest aphidicidal activity, with the 24 h mortality rate of 52.98%, which was slightly lower than the positive control (pyrethrin II, 83.52%).
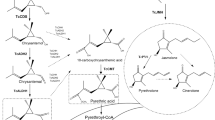
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In agriculture, pests can adversely affect the quality and quantity of the crops production [1]. Currently, pests control mainly relies on the use of chemical synthetic pesticides [2]. Although chemical synthesis pesticides can bring significant benefits to agricultural production in a short time, it is easy to cause environmental pollution and pesticide chemical residues of pesticides [3, 4]. Therefore, it is worthwhile to find and develop ecologically safe pesticides. Previous studies have found that many plant-derived pesticides, including pyrethrin, marine and rotenone, are low toxicity, safe, efficient and easily degradable [5,6,7,8].
Pyrethrins from Pyrethrum cinerariifolium Trev., which are mainly composed of six compounds (pyrethrin I and pyrethrin II, cinerin I and cinerin II, jasmolin I and jasmolin II) with similar structures, are representative excellent insecticidal chemicals [9, 10]. Pyrethrins have tactile toxicity to many agricultural pests, including aphids, weevils, mosquitoes and thrips, by acting on Na+ channels in the insect nervous system [11,12,13,14]. In addition, pyrethrins have a short half-life of about 2 h and don’t leave toxic residues in the environment, so it is commonly recognized as an environmentally friendly pesticide [15]. To find out more chemical constituents of pyrethrins with insecticidal activity, we conducted further phytochemical studies on P. cinerariifolium seeds. In this research, eight pyrethrins (Fig. 1) were identified, including four undescribed compounds and four known compounds, and their insecticidal activities were evaluated.

Structures of compounds 1–8
2 Results and discussion
2.1 Structural identification of compounds
Pyrethrin C (1) was a colorless oil. The HRESIMS ion peak at m/z 305.1395 ([M‒H]‒, calcd for 305.1394) indicated its molecular formula as C17H22O5. The characteristic signals for protons were observed in 1H NMR spectrum (Table 1), included three methyl groups (δH 1.09, s; δH 1.36, s; δH 2.11, s), a methylene group (δH 2.26, dd, J = 18.8, 2.3 Hz; δH 2.93, dd, J = 18.8, 6.3 Hz), and three methine groups (δH 2.89, d, J = 5.2 Hz; δH 2.45, d, J = 5.2 Hz; δH 5.65, d, J = 6.3 Hz). Besides, the 13C and DEPT NMR spectra showed 17 carbon signals, attributed to carbons of four methyls, three methylenes (an olefinic, an oxygenated and an aliphatic methylenes), and three methines (an oxygenated and two aliphatic methine), as well as seven non-protonated carbons (three olefinic and three carbonyls). The aforementioned information proved that compound 1 was similar to the 4(S)-1-oxo-2-allyl-3-methyl-2-cyclopente-4-yl-2,2-dimethyl-3(R)-(2-methyl-1-propenyl)-1(R)-cyclopropane carboxylate [16]. The main differences between them were that the C-7 (δC 196.9) was a carbonyl instead of a methylene, and the vinylene attached to the C-7ʹ was replaced by the OH group in 1D NMR spectra of 1. The above inference can be unambiguously verified by the HMBC cross-peaks (Fig. 2) of H-9a (δH 6.02), H-10 (δH 1.90), H-3 (δH 2.45), and H-1 (δH 2.89) with the carbonyl (C-7); of H-7ʹ (δH 4.38) with C-3ʹ (δC 141.5), C-4ʹ (δC 205.1) and C-6ʹ (δC 14.0). In the ROESY spectra (Fig. 3), the correlations of H-1 and H-6, H-3 and H-5 indicated the protons of H-1 was β-oriented and H-3 was α-oriented. Moreover, the ROESY correlations of H-6ʹ with H-5ʹb, H-7ʹ, H-1ʹ, and H-1ʹ with H-5ʹb indicated that these protons were co-facial and arbitrarily assigned as β-orientation, as well as H-5ʹa was α-orientation. Meanwhile, the analysis of reported pyrethrins combined with biosynthetic pathways could confirm that H-1ʹ was generally β-orientation [9, 17, 18]. In addition, the ROESY correlations of H-1 with H-7 and H-3 and H-10 manifested that Δ7,8-double bond was E-configuration. The above ROESY correlations and the experimental ECD spectra (Fig. 4) indicated the stereotypic configuration of (1R, 3R, and 1ʹS), consistent with known pyrethrin II in previous studies [17, 19]. Therefore, the structure of 1 was determined and named pyrethrin C.

Key HMBC (blue arrows) and 1H-1H COSY (red line) correlations of 1, 2, 3, and 4

Key ROESY (red dotted arrows) correlations of 1, 2, 3, and 4

Experimental ECD spectrum of pyrethin II, 1, 2, and 3
Pyrethrin D (2) was a yellow oil, and its molecular formula was identified as C22H28O6 through HRESIMS ion at m/z 387.1395 ([M‒H]‒, calculated. 387.1394) with nine degrees of unsaturation. Carefully analyze the 1D NMR spectroscopic data (Table 2) can be found in the similarity of 2 and pyrethrin II (6) [19]. Compared with pyrethrin II (6), the main differences of 2 were that the positions of conjugate double bonds, which transferred from C-8ʹ/C-9ʹ and C-10ʹ/C-11ʹ to C-7ʹ/C-8ʹ and C-9ʹ/C-10ʹ, and the terminal carbon C-11ʹ was attached to a hydroxyl group, which was proved by the long-range 1H-1H COSY correlations (Fig. 2) of H-7/H-8/H-9/H-10/H-11. Simultaneously, the above deduction also could be proved by the key HMBC cross-peaks (Fig. 2) from H-7ʹ (δH 6.21) to C-2ʹ (δC 164.2), C-3ʹ (δC, 137.6), C-4ʹ (δC 202.9), and C-8ʹ (δC 135.1); H-9ʹ (δH 6.34) to C-8ʹ (δC 135.1), C-10ʹ (δC 135.7), and C-11ʹ (δC 63.2); H-11ʹ (δH 4.26) to C-10ʹ (δC 135.7). Besides, according to the 1H NMR data (Table 1), the large value of the coupling constant J7'-8' (15.7 Hz > 15.0 Hz) and J9'-10' (15.3 Hz > 15.0 Hz) suggested their E-configurations. In addition, the ROESY correlations (Fig. 3) of H-1 with H-7 and H-3 with H-10 manifested that Δ7,8-double bond was E-configuration. Upon carefully analyzing the ROESY spectra correlations (H-1 with H-6, H-3 with H-5, H-6ʹ with H-5ʹb, H-7ʹ, H-1ʹ, H-1ʹ with H-5ʹb), and combined with the biosynthesis pathway and the experimental ECD spectra (Fig. 4), which indicated that 2 had the same stereoscopic configuration with pyrethin II (6). The above information indicated that the structure of pyrethrin D (2) as shown in Fig. 1.
Pyrethrin E (3) was obtained as a yellow oil and its molecular formula was identified as C22H28O6 (nine degrees of unsaturation) in terms of HRESIMS data at m/z 387.1817 ([M‒H]‒, calcd for 387.1813). Comparing the 1H and 13C NMR data, it could be found that the structure of compound 3 was similar to the isopyrethrin II [20], except the absence of one double-bonded between C-9ʹ and C-10ʹ in 3, which was replaced by the carbonyl (δC 201.6) located at C-9ʹ. Supporting evidence was discovered in the 2D NMR, the HMBC correlations (Fig. 2) of C-9ʹ (δC 201.6) with H-7ʹ (δH 7.24), H-8ʹ (δH 7.43), H-10ʹ (δH 2.64), H-11ʹ (δH 1.14), and C-8ʹ (δC 130.8), of C-10ʹ (δC 35.9) with H-8ʹ (δH 7.43) and H-11ʹ (δH 1.14), combined with the 1H-1H COSY correlation (Fig. 2) of H-10ʹ and H-11ʹ, confirmed the aforementioned deduction. The ROESY correlations (Fig. 3) of H-1 with H-7 and H-3 with H-10 manifested that Δ7,8-double bond was E-configuration. In addition, the ROESY correlations (of H-1 with H-6, H-3 with H-5, H-6ʹ with H-5ʹb, H-7ʹ, H-1ʹ, H-1ʹ with H-5ʹb) and the same ECD spectra (Fig. 4) indicated 3 had the same stereoscopic configuration with pyrethin II (6). Thus, the structure of 3 was established and named pyrethrin E.
At the same time, the experimental ECD spectra (Fig. 4) further proved that compounds 1, 2, 3 and pyrethin II (6) had the same Cotton effect, which can be determined that their stereoscopic configuration was consistent.
Compound 4 was isolated as a yellow oil. It had a molecular formula of C25H34O7 based on ion peak at m/z 469.2197 ([M + Na]+, calcd for 469.2201) as given by HRESIMS data. Meanwhile, there were nine degrees of unsaturation in 4. A detailed comparison of the 1H and 13C NMR spectroscopic data (Tables 1, 2) of 4 with the 10ʹ,11ʹ-dihydroxypyrethrin II [18] revealed that there were three additional carbon signals including two methyl signals (δC 25.9, C-13ʹ; δC 26.8, C-14ʹ) and a quaternary carbon signal (δC 109.2, C-12ʹ) indicated an O-isopropyl motif was presented in 4. The HMBC correlations (Fig. 2) from H-13ʹ (δH 1.43), H-14ʹ (δH 1.41) to C-12ʹ (δC 109.2) determined the position of O-isopropyl motif. The planar structure of 4 was thereby finally established. The ROESY correlations (Fig. 3) of H-1 with H-7, H-3 with H-10 manifested that Δ7,8-double bond was E-configuration. In addition, the correlations of H-1 with H-6, H-3 with H-5, H-6ʹ with H-5ʹb, H-7ʹ, H-1ʹ, H-1ʹ with H-5ʹb in ROESY spectrum indicated that the relative configuration of H-1, H-3 and H-1ʹ were consistent with 1. Thus, H-1 and H-1ʹ were identified as β-orientation and H-3 was α-orientation. However, the stereoscopic configuration of H-10ʹ could not be determined by the ROESY correlation, the ECD calculations were performed to determine it. Finally, the consistency between the calculated ECD result of (1R, 3R, 1ʹS, and 10ʹS) and the experimental ECD (Fig. 5) determined the absolute stereochemistry of pyrethin F (4).

Experimental ECD spectrum of 4 and calculated ECD spectrum of (1R,3R,1′S,10′S)-4 and (1R,3R,1′S,10′R)-4
The identification of other known compounds, including pyrethin I (5), pyrethin II (6), cinerin II (7) and jasmolin II (8), were determined by comparison their 1D NMR data with the reported compounds [19].
2.2 Insecticidal activity
The aphidicidal activities of compounds 1–4 were evaluated at a concentration of 0.1 mg/mL. The results (Table 3) showed that the 24 h mortality rate of compounds 1, 2, 3, and 4 ranged from 17.64% to 52.94%, which were slightly lower than the mortality rate of the positive control (pyrethin II, 83.52%). 2 and 4 showed the moderate activity (52.94% and 41.17%), indicating that they could be used as one of the active components of naturally obtained insecticides.
Based on the above activity data, the structure–activity relationships were preliminarily discussed. Compared with the positive control (pyrethrin II), the activities of the compounds 1–4 decreased when the side chain structure of the three-membered ring or five-membered ring changed. Compounds with conjugated double bonds in C-3ʹ side chains of five-membered rings, such as 2 and pyrethrin II, showed better insecticidal activity than 1, 3 and 4. According to the higher activity of 2 compared with 3, carboxyl substitution on C-9ʹ might decrease the aphidicidal activity.
3 Experimental section
3.1 General experimental procedure
Fractions were examined by TLC on silica gel GF254 plates (200–250, Qingdao Marine Chemical, Inc.), and the 10% H2SO4 in ethanol as developer. The silica gel (200–300 mesh, Qingdao Marine Chemical, Inc., Qingdao, China), reversed-phase C18 silica gel (40–60 μm, Merck, Darmstadt, Germany), and Sephadex LH-20 (Pharmacia, Stockholm, Sweden) were used as the material for column chromatography (CC) analysis. The Agilent 1100 or 1260 liquid chromatography system equipped with Agilent ZORBAX SB-C18 columns (5 μm, 4.6 × 250 mm) was used for HPLC analysis. 1D and 2D NMR spectra were obtained using the Bruker AV-600 and AV-800 spectrometers (Bruker, Zürich, Switzerland) with tetramethylsilane (TMS) as an internal standard. The Agilent UPLC system spectrometer (Agilent Technologies, Foster City, CA, USA) was used to obtain HRESIMS data. The Rudolph Autopol VI polarimeter (Hackettstown, NJ, USA) was used to obtain optical rotations. UV spectra were detected on an UV-2401 PC spectrometer (Shimadzu Corp., Japan). Acyrthosiphon pisum was obtained from Henan Quanying Insect Biology Co., LTD (Henan, China).
3.2 Plant materials
The seeds of Pyrethrum cinerariifolium Trev. were collected in Xinjiang Province, People’s Republic of China in September 2020. The plant was authenticated by Mr. Zhong-Rong Li Senior Engineer, Kunming Institute of Botany, Chinese Academy of Sciences. The specimen (KUN. No. Q20200915) was deposited in the State Key Laboratory of Photochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and isolation
The dried P. cinerariifolium seeds (11.5 kg) were extracted with 90% acetone (25 L × 3) at room temperature. The residue (1.1 kg) which was obtained after the acetone solvent was removed by a rotary evaporator, was mixed with appropriate amount of H2O, and then extracted with petroleum ether (PE, 10 L × 3) and ethyl acetate (EtOAc, 10 L × 3). Then, the EtOAc part (314.9 g) was separated by silica gel CC eluted with PE/EtOAc (20:1, 10:1, 1:1, 1:10, 1:20, 1:40, and 1:50) to obtain seven fractions (Fr.I–Fr.VII).
Fr.II (5 g) was separated by silica gel CC (PE/EtOAc, 15:1, 12:1, 10:1, 8:1, 5:1) to give five fractions (Fr.II-1–Fr.II-5). Fr.II-2 (56 mg) was further purified by the semipreparative HPLC with same gradient elution to give 5 (84% MeCN/H2O, 6.4 mg, tR = 17.6 min) and 8 (57% MeCN/H2O, 7.1 mg, tR = 44.2 min).
The separation of Fr. III (39 g) was firstly carried out using an RP-18 column eluted with the MeOH/H2O (50:50, 60:40, 70:30, and 75:25), and produced six fractions (Fr. III-1–Fr. III-6). Then, Fr.III-2 (5 g) was further separated by Sephadex LH-20 column to get three fractions (Fr. III-2-1–Fr. III-2-3), in which MeOH was used as eluent. After that, all subfractions were further separated by a silica gel CC with CH2Cl2-EtOAc (15:1, 12:1, 10:1, 7:1, 5:1, 3:1, and 1:1) and purified by semipreparative HPLC eluted with CH3OH/H2O or MeCN/H2O to afford compounds 1 (48% CH3OH/H2O, 1.9 mg, tR = 30.1 min), 6 (65% MeCN/H2O, 34.0 mg, tR = 27.5 min), and 7 (85% MeCN/H2O, 5.2 mg, tR = 21.2 min).
Fr. IV (29.0 g) was treated into five sub-fractions (Fr.IV-1–Fr.IV-5) using RP-18 column eluting sequentially with the solvents system of MeOH/H2O (30:70 to 70:30). Then, Fr.IV-1 (2 g) was fractionated into four subfractions (Fr.IV-1-1–Fr.IV-1-4) using Sephadex LH-20 column with MeOH. Fr.IV-1-3 (52 mg) was purified by semipreparative HPLC eluted with MeCN/H2O to give compound 3 (46% MeCN/H2O, 10.5 mg, tR = 34.1 min). Fr.IV-3 (10 g) was further separated by silica gel CC (CH2Cl2-EtOAc, 10:1, 7:1, 5:1, 3:1, 1:1, 1:3, and 1:5) to give 12 subfractions (Fr.IV-3-1–Fr.IV-3-12). Fr.IV-3-5 (112 mg) was purified by semipreparative HPLC eluted with CH3CN/H2O to give compound 2 (32% MeCN/H2O, 1.5 mg, tR = 36.8 min) and 4 (52% MeCN/H2O, 2.3 mg, tR = 28.9 min).
3.4 Compound characterization
PyrethrinC (1): colorless oil; C17H22O5; [α] 21D ‒14.6 (c 0.11, CH3OH); UV (MeOH) λmax (logε): 309 (2.82) and 222 (3.91) nm; HRESIMS m/z 305.1395 [M–H]‒ (calcd for. 305.1394, C17H21O5).
Pyrethrin D (2): light yellow oil; C22H28O6; [α] 21D ‒20.0 (c 0.12, CH3OH); UV (MeOH) λmax (logε): 238 (4.04) and 196 (3.84) nm; HRESIMS m/z 387.1395 [M–H]‒ (calcd for. 387.1394, C22H28O6).
Pyrethrin E (3): light yellow oil; C22H28O6; [α] 21D ‒33.64 (c 0.11, CH3OH); UV (MeOH) λmax (logε): 257 (4.30) and 197 (4.11) nm; HRESIMS m/z 387.1817 [M–H]‒ (calcd for. 387.1813, C22H28O6).
Pyrethrin F (4): light yellow oil; C25H34O7; [α] 21D ‒28.4 (c 0.10, CH3OH); UV (MeOH) λmax (logε): 233 (4.24) and 197 (3.97) nm; HRESIMS m/z 469.2197 [M + Na]+ (calcd for. 469.2201, C25H34O7).
The 1H NMR data were shown in Table 1, 13C NMR data were shown in Table 2, and 2D NMR data (HSQC, HMBC, 1H-1H COSY, ROESY) were shown in Additional file 1 for pyrethrin (C–F).
3.5 Bioassay
The contact toxicity assay was carried out according to the previous method with some modifications [21]. The same-size A.pisum adults were collected into the insect culture box with fresh pea seedlings and moist tissue paper for experimentation. The insect culture box was sealed with plastic wrap and left holes for air permeability. The test samples were accurately measured and dissolved in acetone to prepare sample solutions of 0.1 mg/mL. Then 2 μL of the diluted acetone solutions were applied to the dorsal thorax of the A.pisum adults. 30 A.pisum adults were used for each group, and each experiment was replicated three times. The A.pisum adults of each treatment were transferred to the corresponding insect culture box. The mortality was evaluated after 24 h and 48 h. If the A.pisum could not move when disturbed by a wet brush, they were considered dead. Pyrethrin II was used as a positive control, and acetone treatments were determined as a blank control. Mortality was corrected by Abbott's formula [22].
3.6 ECD calculations
The experimental ECD spectra of the compounds were recorded in MeOH. The ECD calculation was performed as previously reported [23]. The specific calculation process and data were described in the Additional file 1.
4 Conclusion
In summary, eight pyrethrins were isolated from seeds of P. cinerariifolium, including four new compounds (1–4) and four known compounds (5–8). All of them had the same core structure as pyrethrins, in which the side chain on C-3 and C-3ʹ of pyrethrin C (1) was changed, and the side chain on C-3ʹ of pyrethrin D (2), pyrethrin E(3), and pyrethrin F (4) were mainly changed. Meanwhile, we also tested the aphidicidal activities of 1, 2, 3, and 4, and the results exhibited that they all had aphidicidal activity, among which pyrethrin D (2) had the strongest mortality rate of 52.94%. These findings suggested that the P. cinerariifolium can be used as an important source of the insecticidal ingredient and continue to develop insecticides for agricultural production.
Availability of data and materials
The data that support the findings of this study were available on request from the corresponding author, upon reasonable request.
References
Liu W, Liu Z, Huang C, Lu M, Liu J, Yang Q. Statistics and analysis of crop yield losses caused by main disease and insect pests in recent 10 years. Plant Prot. 2016;42:1–9.
Hu J, Wang W, Dai J, Zhu L. Chemical composition and biological activity against Tribolium castaneum (Coleoptera: Tenebrionidae) of Artemisia brachyloba essential oil. Ind Crop Prod. 2019;128:29–37.
Liu JL, Yu JF, Yin JL, Wu JC. Research progress on the effect of chemical pesticides on plant physiology and biochemistry. Agrochemicals. 2006;45:511–4.
Ma YN, Xu FR, Chen CJ, Li QQ, Wang MZ, Cheng YX, Dong X. The beneficial use of essential oils from buds and fruit of Syzygium aromaticum to combat pathogenic fungi of Panax notoginseng. Ind Crop Prod. 2019;133:185–92.
Davies TGE, Field LM, Usherwood PNR, Williamson MS. DDT, pyrethrins, pyrethroids and insect sodium channels. IUBMB Life. 2007;59:151–62.
Khan MS, Akbar MF, Sultan A, Saleem MS, Gul C. Testing the effect of different insecticides on Myzus persicae (Homoptera: Aphididae) in field mustard (Brassicae campestris L.) Czern for possible consideration in an IPM strategy. Int J trop Insect Sci. 2020;40:225–31.
Wu JH, Yu XT, Wang XS, Tang LD, Shaukat A. Matrine enhances the pathogenicity of Beauveria brongniartii against Spodoptera litura (Lepidoptera: Noctuidae). Front Microbiol. 2019;10:1812.
Zhang PW, Qin DQ, Chen JJ, Zhang ZX. Plants in the Genus Tephrosia: valuable resources for botanical insecticides. Insects. 2020. https://doi.org/10.3390/insects1110072.
Jeran N, Grdiša M, Varga F, Šatović Z, Liber Z, Dabić D, Biošić M. Pyrethrin from Dalmatian pyrethrum (Tanacetum cinerariifolium (Trevir.) Sch. Bip.): biosynthesis, biological activity, methods of extraction and determination. Phytochem Rev. 2021;20:875–905.
Ujihara K. The history of extensive structural modifications of pyrethroids. J Pestic Sci. 2019;44:215–24.
Yang T, Gao LP, Hu H, Stoopen G, Wang CY, Jongsma MA. Chrysanthemyl diphosphate synthase operatesin plantaas a bifunctional enzyme with chrysanthemol synthase activity. J Biol Chem. 2014;289:36325–35.
Andreev R, Kutinkova H, Baltas K. Non-chemical control of some important pests of sweet cherry. J Plant Prot Res. 2008;48:503–8.
Chermenskaya TD, Stepanycheva EA, Shchenikova AV, Chakaeva AS. Insectoacaricidal and deterrent activities of extracts of Kyrgyzstan plants against three agricultural pests. Ind Crop Prod. 2010;32:157–63.
Narahashi T. Neuroreceptors and ion channels as the basis for drug action: past, present, and future. J Pharmacol Exp Ther. 2000;294:1–26.
Gunasekara AS. Environmental fate of pyrethrins. Environmental Monitoring Branch. Department of Pesticide Regulation. Sacramento, CA, 2004; 1–19.
Jacques M, Jean T, Demoute, JP. Optically active allethrolone. European Patent Organization. 1981; EP21926-A1.
Kawamoto M, Moriyama M, Ashida Y, Matsuo N, Tanabe Y. Total syntheses of all six chiral natural pyrethrins: accurate determination of the physical properties, their insecticidal activities, and evaluation of synthetic methods. J Org Chem. 2020;85:2984–99.
Freemont JA, Littler SW, Hutt OE, Mauger S, Meyer AG, Winkler DA, Kerr MG, Ryan JH, Cole HF, Duggan PJ. Molecular markers for pyrethrin autoxidation in stored pyrethrum crop: analysis and structure determination. J Agric Food Chem. 2016;64:7134–41.
Lu H, Zhu H, Dong HJ, Guo LP, Ma TY, Wang X. Purification of pyrethrins from flowers of Chrysanthemum cineraraeflium by high-speed counter-current chromatography based on coordination reaction with silver nitrate. J Chromatogr A. 2020;1613: 460660.
Wenclawiak BW, Krappe M, Otterbach A. In situ transesterification of the natural pyrethrins to methyl esters by heterogeneous catalysis using a supercritical fluid extraction system and detection by gas chromatography-mass spectrometry. J Chromatogr A. 1997;785:263–7.
Rubens CZ, Carolina GP, Adélia MB, Matheus B, Thaisa SL, Edson JM, Alessandra B, Carlos ENM. Insecticidal and antifungal activities of Melaleuca rhaphiophylla essential oil against insects and seed-borne pathogens in stored products. Ind Crop Prod. 2022;182: 114871.
Abbott W. A method of computing the effectiveness of an insecticide. J Econ Entomol. 1925;18:265–7.
Chen XH, Liu X, Cui WB, An FL, Liu L, Wu Q, Yu JN, Dai JY, Zhang ZX, Fei DQ. Highly oxygenated germacrane-type sesquiterpenoids from the whole plant of Salvia cavaleriei H.Lév. and their biological activities. Phytochem. 2023;211: 113686.
Acknowledgements
The authors gratefully acknowledge the financial support of this study by the Key Research and Development Program of Yunnan Province, China (202003AD 150006), as well as the Cooperation Project with DR PLANT Company (2023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
We state that we have no known competing financial interests or personal relationships that could affect the work reported in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional
file 1: Figure S1. 1H NMR spectrum (800 MHz) of compound 1 in CD3Cl. Figure S2. 13C NMR spectrum (200 MHz) of compound 1 in CD3Cl. Figure S3. HSQC spectrum of compound 1 in CD3Cl. Figure S4. HMBC spectrum of compound 1 in CD3Cl. Figure S5. 1H-1H COSY spectrum of compound 1 in CD3Cl. Figure S6. ROESY spectrum of compound 1 in CD3Cl. Figure S7. HRESI (-) MS spectrum of compound 1. Figure S8. OR of compound 1. Figure S9. UV spectrum of compound 1. Figure S10. 1H NMR spectrum (600 MHz) of compound 2 in CD3Cl. Figure S11. 13C NMR spectrum (150 MHz) of compound 2 in CD3Cl. Figure S12. HSQC spectrum of compound 2 in CD3Cl. Figure S13. HMBC spectrum of compound 2 in CD3Cl. Figure S14. 1H-1H COSY spectrum of compound 2 in CD3Cl. Figure S15. ROESY spectrum of compound 2 in CD3Cl. Figure S16. HRESI (-) MS spectrum of compound 2. Figure S17. OR of compound 2. Figure S18. UV spectrum of compound 2. Figure S19. 1H NMR spectrum (600 MHz) of compound 3 in CD3Cl. Figure S20. 13C NMR spectrum (150 MHz) of compound 3 in CD3Cl. Figure S21. HSQC spectrum of compound 3 in CD3Cl. Figure S22. HMBC spectrum of compound 3 in CD3Cl. Figure S23. 1H-1H COSY spectrum of compound 3 in CD3Cl. Figure S24. ROESY spectrum of compound 3 in CD3Cl. Figure S25. HRESI (-) MS spectrum of compound 3. Figure S26. OR of compound 3. Figure S27. UV spectrum of compound 3. Figure S28. 1H NMR spectrum (800 MHz) of compound 4 in CD3Cl. Figure S29. 13C NMR spectrum (200 MHz) of compound 4 in CD3Cl. Figure S30. HSQC spectrum of compound 4 in CD3Cl. Figure S31. HMBC spectrum of compound 4 in CD3Cl. Figure S32. 1H-1H COSY spectrum of compound 4 in CD3Cl. Figure S33. ROESY spectrum of compound 4 in CD3Cl. Figure S34. HRESI (+) MS spectrum of compound 4. Figure S35. OR of compound 4. Figure S36. UV spectrum of compound 4. Figure S37. Five optimized conformers of 4-1. Table S1. Conformational analysis of the eight optimized conformers of 4-1 in the gas phase (T = 298.15 K). Table S2. Atomic coordinates (Å) of 4-1a obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S3. Atomic coordinates (Å) of 4-1b obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S4. Atomic coordinates (Å) of 4-1c obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S5. Atomic coordinates (Å) of 4-1d obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S6. Atomic coordinates (Å) of 4-1e obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Figure S38. Seven optimized conformers of 4-2. Table S7. Conformational analysis of the eight optimized conformers of 4-2 in the gas phase (T = 298.15 K). Table S8. Atomic coordinates (Å) of 4-2a obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S9. Atomic coordinates (Å) of 4-2b obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S10. Atomic coordinates (Å) of 4-2c obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S11. Atomic coordinates (Å) of 4-2d obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S12. Atomic coordinates (Å) of 4-2e obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S13. Atomic coordinates (Å) of 4-2f obtained at the CAM-B3LYP/TZVP level of theory in the MeOH. Table S14. Atomic coordinates (Å) of 4-2g obtained at the CAM-B3LYP/TZVP level of theory in the MeOH.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, HR., Lin, LW., Li, ZR. et al. Four undescribed pyrethrins from seeds of Pyrethrum cinerariifolium and their aphidicidal activity. Nat. Prod. Bioprospect. 13, 22 (2023). https://doi.org/10.1007/s13659-023-00385-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-023-00385-0