
Abstract
The parallel sequencing of targeted amplicons is a scalable application of next-generation sequencing (NGS) that can advantageously replace Sanger sequencing in certain DNA barcoding studies. It can be used to sequence different PCR products simultaneously, including co-amplified products. Here, we explore this approach by simultaneously sequencing five markers (including the DNA barcode and a diagnostic marker of Wolbachia) in 12 species of Halictidae that were previously DNA barcoded using Sanger sequencing. Consensus sequences were obtained from fresh bees with success rates of 74–100% depending on the DNA fragment. They improved the phylogeny of the group, detected Wolbachia infections (in 8/21 specimens) and characterised haplotype variants. Sequencing cost per marker and per specimen (11.43 €) was estimated to decrease (< 5.00 €) in studies aiming for a higher throughput. We provide guidelines for selecting NGS or Sanger sequencing depending on the goals of future studies.
Similar content being viewed by others


1 Introduction
DNA barcoding is a standardised and widely used method to identify specimens at the species level using a restricted set of short DNA fragments—usually only the 5’ end of the cytochrome c oxidase subunit I (COI) in animals—(Hebert et al. 2003). The standard DNA barcoding protocol relies on Sanger sequencing, but next-generation sequencing (NGS) technologies can improve or complement the standard DNA barcoding pipeline (Shokralla et al. 2014, 2015; Batovska et al. 2017; Wilkinson et al. 2017; Hebert et al. 2018). These methods, referred to as “next-generation DNA barcoding” (Shokralla et al. 2014) or “targeted amplicon sequencing” (Bybee et al. 2011), enable the analysis of mixtures of DNA fragments that are co-amplified during PCR or obtained by pooling different PCR products. In insect systematics, these methods can be profitably used to (1) sequence multiple loci at relatively reduced costs, (2) improve single gene phylogenies and (3) assess the presence of cytoplasmic endosymbiotic bacteria such as Wolbachia (Breeuwer and Werren 1993; James et al. 2002; Hiroki et al. 2004; Raychoudhury et al. 2010). These bacteria are frequently detected in Halictidae and can affect the transmission of the mitochondrial genome (Smith et al. 2012). They can also be used to detect variants in the PCR products that can be due to heterozygosity, heteroplasmy or nuclear copies of COI (nuclear mtDNA = numts) (Buhay 2009). All these issues can affect gene trees in Hymenoptera (Magnacca and Brown 2010; Cristiano et al. 2012).
Here, we implemented the parallel sequencing of targeted amplicons to (1) re-sequence the COI barcode fragment, (2) sequence three nuclear gene fragments and (3) sequence a fragment of the Wolbachia outer surface protein gene in 12 Halictid species that were recently studied by DNA barcoding using Sanger sequencing (Pauly et al. 2015). These species belong to Halictus (Seladonia) Robertson, 1918 [or Seladonia depending on its assignment as a subgenus (Michener 2007) or genus (Pesenko 1999, 2004)] and include five species belonging to the H. smaragdulus Vachal 1895 [or S. smaragdula] species complex. While COI data strongly supported the delineation of these five species, they did not fully resolve the phylogenetic relationships of the group (Pauly et al. 2015). The present small-scale NGS implementation explores to what extent NGS can effectively contribute to solve the aforementioned issues.
2 Material and methods
2.1 Sampling and DNA sequencing
We sampled 21 specimens (Table I) representing five of the six species of the Halictus smaragdulus complex and seven closely related Halictidae species showing the smallest interspecific p-distances at COI with respect to the complex (Pauly et al. 2015). One species of the complex H. cretellus (Pauly and Devalez 2015 in Pauly et al. 2015) is only known from Crete (Pauly et al. 2015; Schmidt et al. 2015) and could not be sampled for this study. Most specimens were collected after 2011 and were captured with a net, killed with ethyl acetate and stored in absolute ethanol. Two specimens date back > 40 years ago (AP030 in 1973 and AP048 in 1890). Genomic DNA was extracted from one middle leg using the NucleoSpin Tissue Kit (Macherey-Nagel, Germany). We targeted five gene fragments; four of them were used for phylogenetic tree reconstructions and included COI and three nuclear markers that were previously used for phylogenetic analysis in hymenopterans, viz. wingless (wnt1), white (w) and a hippo gene (HOG7036-02) for putative serine/threonine kinase, exons 1-2 (Danforth et al. 2004; Kawakita et al, 2008; Gibbs et al. 2012; Hartig et al, 2012). COI was sequenced and used for both phylogenetic reconstruction and assessing the presence of COI pseudogenes, heteroplasmy and Wolbachia COI. Finally, a gene fragment of the Wolbachia outer surface protein (wsp) was used to assess the presence of Wolbachia.
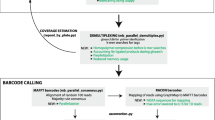
The library preparation (Figure 1a) consisted of a two-step PCR approach (Cruaud et al. 2017). A first round of PCR was performed to amplify six DNA fragments from the five targeted genes (including two overlapping fragments for COI). The Multiplex PCR Kit (QIAGEN, The Netherlands) was used to amplify one to three fragments per reaction (Figure 1a) using tailed forward and reverse primers (TAG1 and TAG2, respectively—Table II). PCR products were purified using AMPure XP beads (Agencourt Biosciences, USA) with a volume ration of 1:1, then diluted to 10 ng/μl and used as template in a second PCR with primers consisting of TAG1 and TAG2, a molecular identifier (MID) of six nucleotides (in the forward primer only) and the Illumina adapters of the TruSeq Custom Amplicon kit (Illumina, USA). After another purification (volume ratio of the PCR product versus AMPure XP beads of 0.9:1), PCR products were pooled and sequenced in one lane of a MiSeq Sequencing System flow cell (Illumina, USA) using the paired-end protocol of the Reagent Nano Kit v. 2 (2 × 250 bp).

Procedure used for library preparation (a) and for bioinformatic analysis (b).
2.2 Data analysis
MiSeq data were demultiplexed and cleaned using Trimmomatic v. 0.32 (Bolger et al. 2014) with the commands ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 TRAILING:28 LEADING:28 SLIDINGWINDOW:5:25 MINLEN:80. AlienTrimmer v. 0.4.0 (Criscuolo and Brisse 2013) was used to remove remaining PCR primers. Paired-end reads were assembled with PEAR v. 0.9.6 (Zhang et al. 2014) and NextAllele (O’Neill et al. 2013) was used to identify the reads obtained for each targeted fragment and to get the consensus sequences (Figure 1b).
Reads obtained for wsp were used to identify Wolbachia haplotypes using the Wolbachia wsp typing module of the Wolbachia multilocus sequence typing (MLST) system (Baldo et al. 2006), a central depository of Wolbachia bacterial and host information (Jolley and Maiden 2010). Heteroplasmy (for COI), heterozygosity (for nuclear genes) and undesired co-amplified products (paralogues or contaminants) were investigated using assemblies with sequencing depth (number of reads per position) > 20. For these assemblies, we calculated the average rate of substitution per base. Geneious v. 10.2.3 (Kearse et al. 2012) was used to examine all variant nucleotides showing a frequency > 10%, a value known to be much higher than sequencing error rates reported for different DNA library preparations and sequencing with the Illumina (Illumina, USA) platform (Schirmer et al. 2016).
Phylogenetic analyses were conducted on different datasets in order to compare topologies and resolutions obtained with the different gene fragments: COI (21 specimens, 658 bp), wnt1 (14, 383 bp), w (21, 384 bp), HOG7036-02 (19, 417 bp), the concatenation of the three nuclear fragments (17, 1184 bp) and the four fragments (17, 1842 bp). In order to assess the added value of including nuclear fragments to a COI phylogeny, we compared the topologies of the COI dataset including only specimens used in the concatenated datasets (17, 658 bp) with that of the concatenated dataset. Unique haplotypes were extracted using the R packages ape (Paradis et al. 2004) and pegas (Paradis 2010). When alternative haplotypes were observed for the same individual, phylogenetic analyses were repeated with the different haplotypes (instead of using the consensus sequences). Sequences of two outgroup taxa, one Halictidae, Dufourea novaeangliae (Robertson 1897), and one Apidae, Apis mellifera Linnaeus 1758, were retrieved from GenBank (Table I). Neighbour-joining trees were constructed in MEGA 7.026 (Kumar et al. 2016) using uncorrected p-distances and with pairwise deletion and 1000 bootstrap pseudo-replicates. Maximum parsimony (MP) trees were searched using the R package phangorn (Schliep 2011), using the parsimony ratchet heuristic method (Nixon 1999), with characters of equal weights, gaps considered as missing data and using 500 non-parametric bootstrap replicates. For Bayesian phylogeny inference (BI), best partition scheme and best-fit substitution models were estimated using PartitionFinder v. 1.1 (Lanfear et al. 2014) on the basis of seven partitions: one partition for each codon position of COI, one partition for wingless, one partition for w, one partition for the two exons of HOG7036-02 and one partition for the intron of HOG7036-02 (the latter gene fragments were too short to be partitioned according to codon position). BI analyses were performed with MrBayes v. 3.2.6 (Ronquist et al. 2012) and two parallel runs with four chains each were run for five million generations, with unlinked nucleotide substitution parameters for each data partition. Every 1000th generation was sampled, and the first 25% of the trees were discarded (“burn-in”). Convergence was monitored and average standard deviation of split frequencies was < 0.01 after five million generations. Analyses using the maximum likelihood (ML) method were conducted using RAxML (Stamatakis 2015) on the CIPRES Science Gateway (Miller et al. 2010) with 1000 bootstrap pseudo-replicates and the same partition scheme as the for the BI.
3 Results
3.1 Data collection
Overall, 260,214 reads (paired and unpaired) were assigned to the targeted gene fragments (Figure 2). Read quality scores (Phred) ranged from 28 to 40 (mean values between 38 and 39 depending on the specimens). The average rate of substitution per base varied from 0.002 to 0.004 depending on the assemblies. Numbers of reads per specimen obtained for each DNA fragment (Table I) ranged from zero (for the two old museum specimens) to 21,336 reads (for w in AP031). The COI consensus sequences were identical to the COI sequences obtained by Sanger sequencing (Pauly et al. 2015) when sequencing depth was ≥ 5. Hence, we discarded consensus sequences when sequencing depth was < 5. For fresh specimens, we obtained haplotypes with success rates of 100% for COI (aligned length of 658 bp) and w (385 bp), 95% for HOG7036-02 (417 bp) and 74% for wnt1 (383 bp). Concerning the older museum specimens collected in 1890 (AP048) and 1973 (AP030), sequencing depth was always < 5 except for w of AP048 (Table I). A Wolbachia COI consensus sequence was recovered for specimen AP001. The COI alignment comprised 105 variable sites and showed interspecific p-distances ranging from 2.3 to 12.5%. The nuclear data (wnt1, w and HOG7036-02) comprised 36 variable sites and showed interspecific p-distances ranging from 0 to 2.6%.

Number of reads obtained throughout the bioinformatic analysis.
3.2 Detection of variant haplotypes
Variant nucleotide characters were found in 10 to 50% of the reads of wnt1 (in six specimens), w (one) and COI (three) (Table III). Two variant characters were observed with relative frequencies of 0.45 and 0.50 in wnt1 and w, respectively and within one single specimen (AP027, H. lucidipennis). Other variant characters found in wnt1 with a frequency of 0.11 were situated at the end of the reads (Table III). Finally, the variant characters found in COI occurred in 10–23% of the reads of H. lucidipennis (eight positions) and of both specimens of H. seladonius (17 and 26 positions). Most of them (49/51) corresponded to synonymous substitutions and were observed with a high sequencing depth and in good quality reads. The intra-individual p-distances among these haplotypes were 0.26% for nuclear genes and ≤ 4% for COI (0.2–2.7% within AP027 and 0.2–4.0% within AP055). These values were within the range of interspecific distances measured here (0–2.6% for nuclear genes and 2.3–12.5% for COI). However, these intra-individual distances were always smaller than the distances to the closest heterospecific specimens (> 1.1% for nuclear data and > 7.8% for COI) and the inclusion of these variant haplotypes in the phylogenetic analyses did not affect the trees obtained (all variant grouped in a well-supported cluster). No variant was observed for Wolbachia COI.
3.3 Phylogenetic analyses
The phylogenetic relationships within the H. smaragdulus complex (Figure 3) were fully resolved (with posterior probabilities of one in the BI and bootstrap values > 85 in the ML analysis) using the concatenation of all DNA fragments (COI, wnt1, w and HOG7036-02). Variant haplotypes affected neither the topology, nor the support in the trees. Phylogenies obtained using COI only were slightly less resolved than those obtained using the four gene fragments (Figure 3). Those solely based on nuclear data (both separate and concatenated datasets) only supported a few nodes outside the species complex (Online Resource). The only nodes that were never resolved concerned the relationships among H. seladonius, H. lucidipennis and the clade of H. subauratus and H. subauratoides.

Phylogenies of the Halictus (Seladonia) smaragdulus species complex inferred using a Bayesian inference (BI), maximum likelihood (ML), neighbour-joining (NJ) and parsimony (P) of the concatenated dataset including gene fragments of the cytochrome c oxidase subunit I (COI), wingless (wnt1), white-like (w) and hippo for putative serine/threonine kinase (HOG7036-02). Posterior probabilities > 0.95 (BI) and bootstrap values > 70% (ML, NJ and P) are given at nodes. Support values dropped below these thresholds when only COI was analysed at nodes indicated with a §. Analyses exclusively based on nuclear data are presented as Online Resource. Specimens in which the Wolbachia surface protein gene was detected are tagged with a *.
3.4 Wolbachia infection
Wolbachia sequences of wsp were obtained in eight out of the 21 specimens, with 14 to 831 reads per specimen (Table I). The eight wsp positive specimens belonged to five species (Table I): H. cephalicus (2 detections/2 specimens), H. seladonius (2/2), H. subauratus (1/1), H. smaragdulus (2/2) and H. gemmellus (1/2). All haplotypes queried in the Wolbachia MLST database provided a perfect match with Wolbachia sequences of the supergroup A, a clade of Wolbachia strains commonly found in Hymenoptera (Casiraghi et al. 2005; Ros et al. 2009; Gerth et al. 2011). Five different sequences of the hypervariable region 1 (HVR1) of wsp, coded as numbers 1, 11, 13, 51 and 53, in the Wolbachia MLST database were observed. One or two different HVR1 sequences were detected per specimen. We observed mainly HVR1: 11 in H. cephalicus, HVR1: 51 in H. seladonius and HVR1: 11 and HVR1: 1 in H. smaragdulus (Figure 4). Wolbachia COI was only sequenced in one specimen, AP001, which was also positive for wsp. No exact match was found for this sequence in the MLST database but best matches in GenBank were 99% similar (100% sequence coverage) and mostly (99/100) comprised Wolbachia COI from hymenopterans.

Types of hypervariable region 1 (HVR1) identified in the Wolbachia surface protein (wsp) gene fragment surveyed in this study. Values indicate number of front end reads matching a HVR1 type.
4 Discussion
Parallel sequencing of PCR amplicons is most effective when limited sequence data are targeted per specimen (Mamanova et al. 2010; Grover et al. 2012). This is the case for DNA barcoding or multilocus phylogenetic analyses. Compared to Sanger sequencing, it can improve the sequencing sensitivity (fewer false negatives) and accuracy by enabling the simultaneous detection of co-amplified products such as homologues, paralogues and contaminants (Grover et al. 2012; Shokralla et al. 2014) at relatively reduced costs (Bybee et al. 2011). Below, we evaluate the added value of the protocol applied here compared to standard DNA barcoding using Sanger sequencing.
4.1 Data collection and cost-efficiency
Success rate of parallel amplicon sequencing is expected to highly depend on the PCR amplification. For COI (the only marker that was sequenced both by NGS and Sanger), the usage of NGS did not produce a more complete dataset than with Sanger sequencing since COI could only be obtained from fresh specimens in both cases. The low sequencing depths obtained here for older museum specimens were not considered reliable. The total cost of this analysis (five markers, 21 specimens) was of approximately 1500 € (excluding VAT and labour cost). The cost associated to the NGS implementation (second PCR and the MiSeq sequencing run) was of approximately 1200 € (11.43 € per marker and per specimen). For comparison, sequencing the same PCR products using Sanger sequencing was estimated to cost 546 € (5.2 € per bidirectional read). However, targeting the same number of DNA fragments in 96 samples would become more cost-efficient with NGS (1330 € for 300 Mb output to 1900 € for 7 Gb output) than with Sanger (2496 €). A more uniform molarity of the PCR products and a selection of the Illumina reagent kit in accordance with the number of samples processed can further improve this cost-efficiency. The labour cost was higher (1 person month) than for Sanger data analysis (0.5 person month) but the analysis pipeline developed here can be reused to analyse other projects. On the basis of these estimations, we expect the usage of NGS to be more cost-efficient when more than five markers (DNA fragments < 450 bp) have to be sequenced for more than 100 samples, particularly if several projects using the same approach are planned.
4.2 Detection of variant haplotypes
The average substitution rate per base calculated for each assembly was within the expected range of sequencing error rates reported for amplicon sequencing with the Miseq Illumina platform (Schirmer et al. 2016). They were two orders of magnitude below the threshold of 10% used here to detect variants. Variant haplotypes observed with relative frequencies of 0.45 and 0.50 in two nuclear fragments (wnt1 and w) of one specimen (H. lucidipennis) correspond to heterozygosity. The other variants observed with a frequency of 0.11 at the end of the wnt1 reads more probably correspond to sequencing errors. Indeed, the uneven distribution of sequencing errors along sequencing reads can explain some more frequent sequencing errors (Schirmer et al. 2016). Concerning COI, the reads obtained for three specimens (both specimens of H. seladonius and H. lucidipennis) showed eight to 26 variant nucleotide characters (10–23% of the reads). These variants are not cross-contaminants because they are different from the COI haplotypes sequenced in the other individuals. They are also unlikely numts or sequencing errors because most substitutions (49/51) are synonymous and none are responsible for a stop codon. They are more probably due to heteroplasmy. Heteroplasmy was already reported for Hawaiian Hylaeus (Nesoprosopis) Perkins 1899 (Magnacca and Brown 2010). These variant haplotypes did not affect the phylogenetic trees because both species investigated here (H. seladonius and H. lucidipennis) are relatively divergent from their closest known species. However, the intra-individual divergences observed here (up to 2.7 and 4.0%) are in the range of interspecific divergences in Halictidae (Pauly et al. 2015; Gibbs 2018) and could affect results of DNA barcoding analyses involving closely related species (Magnacca and Brown 2010). Detecting such variants is therefore essential in DNA barcoding. Concerning the detection of numts, we did not observe stop codons or shifts in the reading frame but we cannot totally exclude that nuclear copies were amplified. In this regard, our approach does not offer more guarantees than Sanger sequencing as it also relies on the PCR amplification of small DNA fragments and can be biased by different amplification efficiencies (Cruaud et al. 2017). Sequencing the whole mitochondrial genome represents a better solution to detect numts (Nelson et al. 2012).
4.3 Phylogeny
The lack of resolution of the trees exclusively constructed with nuclear data was not useful to check the species delineation obtained with COI. In contrast, some deeper nodes were only resolved in the analyses combining COI and the three nuclear gene fragments (Figure 3). With this dataset, the two clades identified by morphology (Pauly et al. 2015), viz. (H. phryganicus, H. smaragdulus) and ((H. orientanus, H. submediterraneus) H. gemmellus), were supported in our phylogeny. The Halictidae comprises thousands of species that are often difficult to identify morphologically and whose taxonomy is regularly being refined using COI sequence data. Although COI data provide good support for most morphologically described halictid species (Schmidt et al. 2015), some groups like Lasioglossum (Dialictus) are more problematic (Gibbs 2018). It is therefore useful to consider additional loci or genome skimming (Marcus 2018) both for a better species delineation and for a better understanding of interspecific phylogenetic relationships (Danforth et al. 2013). Obviously, the set of loci analysed here was not useful for species delineation but it clarified the evolutionary history of the species studied.
4.4 Wolbachia infection
The detection of the wsp gene in more than one third of the specimens reveals a high prevalence of Wolbachia in the group under study. Although Wolbachia infections were observed previously for the genus (Gerth et al. 2011), these are the first records for the H. smaragdulus species complex. In five of the eight infected individuals, two different HVR1 sequences were detected. This is also in agreement with previous studies revealing the co-occurrence of more than one Wolbachia sequence type in insects (Breeuwer et al. 1992; Mercot et al. 1995; Perrot-Minnot et al. 1996). We observed the same HVR1 sequence type in conspecific specimens (HVR1:11 in both H. cephalicus and both H. smaragdulus specimens and HVR1:51 in both H. seladonius specimens). Our results confirm that Wolbachia COI can be unintentionally sequenced with PCR primers that are routinely used in Metazoa (Smith et al. 2012) and that a parallel sequencing approach provides good quality results when different DNA fragments are co-amplified.
5 Conclusion
The parallel sequencing of targeted amplicons, as applied here, can advantageously replace DNA barcoding in two cases: when a multilocus dataset has to be assembled for a considerable number of specimens and when variant haplotypes are expected in the sampling. Indeed, our experiment was useful to construct a multilocus dataset consisting of DNA barcodes (COI) and three nuclear gene fragments with a cost-efficiency that is estimated to become interesting compared to Sanger sequencing when more than 100 specimens are investigated. Our experiment also enabled the detection of variant COI haplotypes (with intra-individual divergences in the range of interspecific distances in Halictidae) and mixed sequence types of the intracellular bacteria Wolbachia. This relatively cheap application of NGS may therefore be useful in bee systematics, when these cases are encountered.
References
Abouheif, E., Wray, G.A. (2002) Evolution of the gene network underlying wing polyphenism in ants. Science 297, 249–252
Baldo, L., Hotopp, J.C.D., Jolley, K.A., Bordenstein, S.R., Biber, S.A., Choudhury, R.R., Hayashi, C., Maiden, M.C.J., Tettelin, H., Werren, J.H. (2006) Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 72, 7098–7110
Batovska, J., Cogan, N.O.I., Lynch, S.E., Blacket, M.J. (2017) Using Next-Generation Sequencing for DNA Barcoding: Capturing Allelic Variation in ITS2. G3-Genes Genom. Genet. 7, 19–29
Bolger, A.M., Lohse, M., Usadel, B. (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120
Braig, H.R., Zhou, W., Dobson, S.L., O'Neill, S.L. (1998) Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J. Bacteriol. 180, 2373–2378
Breeuwer, J.A.J., Werren, J.H. (1993) Cytoplasmic incompatibility and bacterial density in Nasonia vitripennis. Genetics 135, 565–574
Breeuwer, J.A.J., Stouthamer, R., Barns, S.M., Pelletier, D.A., Weisburg, W.G., Werren, J.H. (1992) Phylogeny of the cytoplasmic incompatibility microorganism in the parasitoid wasp of the genus Nasonia (Hymenoptera: Pteromalidae) based on 16S ribosomal DNA sequences. Insect. Mol. Biol. 1, 25–36
Buhay, J.E. (2009) “COI-like” sequences are becoming problematic in molecular systematic and DNA barcoding studies. J Crust Biol 29, 96–110
Bybee, S.M., Bracken-Grissom, H.D., Haynes, B.D., Hermansen, R.A., Byers, R.L., Clement, M.J., Udall, J. A, Wilcox, E.R., Crandall, K. A. (2011) Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol. Evol. 3, 1312–23
Casiraghi, M., Bordenstein, S.R., Baldo, L., Lo, N., Beninati, T., Wernegreen, J.J., Werren, J.H., Bandi, C. (2005) Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: Clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology 151, 4015–4022
Criscuolo, A., Brisse, S. (2013) AlienTrimmer: A tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genomics 102, 500–506
Cristiano, M.P., Fernandes-Salomão, T.M., Yotoko, K.S.C. (2012) Nuclear mitochondrial DNA: an Achilles’ heel of molecular systematics, phylogenetics, and phylogeographic studies of stingless bees. Apidologie 43, 527–538
Cruaud, P., Rasplus, J.Y., Rodriguez, L.J., Cruaud, A. (2017) High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy. Sci. Rep. 7, 1–12
Danforth, B.N., Brady, S.G., Sipes, S.D., Pearson, A. (2004) Single-copy nuclear genes recover Cretaceous-age divergences in bees. Syst. Biol. 53, 309–326
Danforth, B.N., Cardinal, S., Praz, C., Almeida, E. A. B., Michez, D. (2013) The impact of molecular data on our understanding of bee phylogeny and evolution. Annu. Rev. Entomol. 58, 57–78
Folmer, O. M, Black, W.H., Lutz, R., Vrijenhoek, R. (1994) DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299
Gerth, M., Geißler, A., Bleidorn, C. (2011) infections in bees (Anthophila) and possible implications for DNA barcoding. Syst. Biodivers. 9, 319–327
Gibbs, J. (2018) DNA barcoding a nightmare taxon: assessing barcode index numbers and barcode gaps for sweat bees. Genome 61, 21–31
Gibbs, J., Brady, S.G., Kanda, K., Danforth, B.N. (2012) Phylogeny of halictine bees supports a shared origin of eusociality for Halictus and Lasioglossum (Apoidea: Anthophila: Halictidae). Mol. Phylogenet. Evol. 65, 926–939
Grover, C.E., Salmon, A., Wendel, J.F. (2012) Targeted sequence capture as a powerful tool for evolutionary analysis. Am. J. Bot. 99, 312–9
Hajibabaei, M., Janzen, D.H., Burns, J.M., Hallwachs, W., Hebert, P.D.N. (2006) DNA barcodes distinguish species of tropical Lepidoptera. Proceedings of the National Academy of Sciences 103 (4):968-971
Hartig, G., Peters, R.S., Borner, J., Etzbauer, C., Misof, B., Niehuis, O. (2012) Oligonucleotide primers for targeted amplification of single-copy nuclear genes in apocritan Hymenoptera. PLoS One 7, e39826
Hebert, P.D.N., Cywinska, A., Ball, S.L., DeWaard, J.R. (2003) Biological identifications through DNA barcodes. Proc. R. Soc. London. Ser. B Biol. Sci. 270, 313–321
Hebert, P.D.N., Penton, E.H., Burns, J.M., Janzen, D.H., Hallwachs, W. (2004) Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA. 101, 14812–14817
Hebert, P.D.N., Braukmann, T.W.A., Prosser, S.W.J., Ratnasingham, S., DeWaard, J.R., Ivanova, N. V., Janzen, D.H., Hallwachs, W., Naik, S., Sones, J.E., Zakharov, E. V. (2018) A Sequel to Sanger: amplicon sequencing that scales. BMC Genomics 19, 219
Hiroki, M., Tagami, Y., Miura, K., Kato, Y. (2004) Multiple infection with Wolbachia inducing different reproductive manipulations in the butterfly Eurema hecabe. Proc. Biol. Sci. 271, 1751–1755
James, A C., Dean, M.D., McMahon, M.E., Ballard, J.W.O. (2002) Dynamics of double and single Wolbachia infections in Drosophila simulans from New Caledonia. Heredity 88, 182–189
Jolley, K.A., Maiden, M.C.J. (2010) BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595
Kawakita, A., Ascher, J.S., Sota, T., Kato, M., Roubik, D.W. (2008) Phylogenetic analysis of the corbiculate bee tribes based on 12 nuclear protein-coding genes (Hymenoptera: Apoidea: Apidae). Apidologie 39, 163–175
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P., Drummond, A. (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649
Kumar, S., Stecher, G., Tamura, K. (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874
Lanfear, R., Calcott, B., Kainer, D., Mayer, C., Stamatakis, A. (2014) Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14, 82
Magnacca, K., Brown, M. (2010) Mitochondrial heteroplasmy and DNA barcoding in Hawaiian Hylaeus (Nesoprosopis) bees (Hymenoptera: Colletidae). BMC Evol. Biol. 10, 174
Marcus, J.M. (2018) Our love-hate relationship with DNA barcodes, the Y2K problem, and the search for next generation barcodes. AIMS Genet. 5, 1–23
Mercot, H., Llorente, B., Jacques, M., Atlan, A., Montchamp-Moreau, C. (1995) Variability within the Seychelles cytoplasmic incompatibility system in Drosophila simulans. Genetics 141, 1015–1023
Michener, C.D. (2007) The Bees of the World. 2nd Edition. Johns Hopkins University Press, Baltimore
Miller, M.A., Pfeiffer, W., Schwartz, T. (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proc. Gatew. Comput. Environ. Work. (GCE), 14 Nov. 2010, New Orleans, LA. pp. 1–8
Nelson, L. A, Lambkin, C.L., Batterham, P., Wallman, J.F., Dowton, M., Whiting, M.F., Yeates, D.K., Cameron, S.L. (2012) Beyond barcoding: a mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae). Gene 511, 131–42
Nixon, K.C. (1999) The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 15, 407–414
O’Neill, E.M., Schwartz, R., Bullock, C.T., Williams, J.S., Shaffer, H.B., Aguilar-Miguel, X., Parra-Olea, G., Weisrock, D.W. (2013) Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Mol. Ecol. 22, 111–129
Paradis, E. (2010) pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–20
Paradis, E., Claude, J., Strimmer, K. (2004) APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290
Pauly, A., Devalez, J., Sonet, G., Nagy, Z.T., Boevé, J.L. (2015) DNA barcoding and male genital morphology reveal five new cryptic species in the West Palearctic bee Seladonia smaragdula (Vachal, 1895) (Hymenoptera: Apoidea: Halictidae). Zootaxa 4034, 257–290
Perrot-Minnot, M.J., Guo, L.R., Werren, J.H. (1996) Single and double infections with Wolbachia in the parasitic wasp Nasonia vitripennis: Effects on compatibility. Genetics 143, 961–972
Pesenko, Y.A. (1999) Phylogeny and Classification of the Family Halictidae Revised (Hymenoptera: Apoidea). J. Kansas Entomol. Soc. 72, 104–123.
Pesenko, Y.A. (2004) The phylogeny and classification of the tribe Halictini with special reference to the Halictus genus-group (Hymenoptera: Halictidae). Zoosyst. Ross. 13, 83–113
Raychoudhury, R., Grillenberger, B.K., Gadau, J., Bijlsma, R., van de Zande, L., Werren, J.H., Beukeboom, L.W. (2010) Phylogeography of Nasonia vitripennis (Hymenoptera) indicates a mitochondrial-Wolbachia sweep in North America. Heredity 104, 318–326
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D.L., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M. A & Huelsenbeck, J.P. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–42
Ros, V.I.D., Fleming, V.M., Feil, E.J., Breeuwer, J.A.J. (2009) How diverse is the genus Wolbachia? Multiple-gene sequencing reveals a putatively new Wolbachia supergroup recovered from spider mites (Acari: Tetranychidae). Appl. Environ. Microbiol. 75, 1036–1043
Schirmer, M., D’Amore, R., Ijaz, U.Z., Hall, N., Quince, C. (2016) Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics 17, 125
Schliep, K.P. (2011) phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593
Schmidt, S., Schmid-Egger, C., Morinière, J., Haszprunar, G., Hebert, P.D.N. (2015) DNA barcoding largely supports 250 years of classical taxonomy: identifications for Central European bees (Hymenoptera, Apoidea partim). Mol. Ecol. Resour. 15, 985–1000
Shokralla, S., Gibson, J.F., Nikbakht, H., Janzen, D.H., Hallwachs, W., Hajibabaei, M. (2014) Next-generation DNA barcoding: using next-generation sequencing to enhance and accelerate DNA barcode capture from single specimens. Mol. Ecol. Resour. 14, 892–901
Shokralla, S., Porter, T.M., Gibson, J.F., Dobosz, R., Janzen, D.H., Hallwachs, W., Golding, G.B., Hajibabaei, M. (2015) Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Sci. Rep. 5, 9687
Smith, M.A., Fisher, B. (2009) Invasions, DNA barcodes, and rapid biodiversity assessment using ants of Mauritius. Front. Zool. 6, 31
Smith, M.A., Bertrand, C., Crosby, K., Eveleigh, E.S., Fernandez-Triana, J., Fisher, B.L., Gibbs, J., Hajibabaei, M., Hallwachs, W., Hind, K., Hrcek, J., Huang, D.W., Janda, M., Janzen, D.H., Li, Y., Miller, S.E., Packer, L., Quicke, D., Ratnasingham, S., Rodriguez, J., Rougerie, R., Shaw, M.R., Sheffield, C., Stahlhut, J.K., Steinke, D., Whitfield, J., Wood, M., Zhou, X. (2012) Wolbachia and DNA barcoding insects: patterns, potential, and problems. PLoS One 7, e36514
Stamatakis, A. (2015) Using RAxML to Infer Phylogenies. Curr. Protoc. Bioinformatics 51, 6.14.1–6.14.14
Ward, P.S. & Downie, D.A. (2005) The ant subfamily Pseudomyrmecinae (Hymenoptera: Formicidae): phylogeny and evolution of big-eyed arboreal ants. Syst. Entomol. 30, 310–335
Wilkinson, M.J., Szabo, C., Ford, C.S., Yarom, Y., Croxford, A.E., Camp, A., Gooding, P. (2017) Replacing Sanger with Next Generation Sequencing to improve coverage and quality of reference DNA barcodes for plants. Sci. Rep. 7, 46040
Zhang, J., Kobert, K., Flouri, T., Stamatakis, A. (2014) PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620
Acknowledgements
Sequencing and library preparation was performed at the Genomics Core of KU Leuven (Belgium) with the help of Sigrun Jackmaert. We would like to thank the valuable suggestions of the two anonymous reviewers.
Funding
This study was funded by the Belgian Science Policy (BELSPO) and supported by the FWO Research Community W0.009.11N’Belgian Network for DNA Barcoding’ (BeBoL).
Author information
Authors and Affiliations
Contributions
GS, ZTN and TB conceived the research and wrote the article. GS and SW collected the data and performed the analyses. AP, ZTN, MV, KJ, JVH and MDM contributed to the interpretation of the data. All authors revised the text and the figures and approved the final manuscript.
Corresponding author
Additional information
Handling editor: Marina Meixner
Utilisation du séquençage de nouvelle génération pour améliorer le codage à barres de l’ADN: leçons tirées d’une étude à petite échelle d’espèces d’abeilles sauvages ( Hymenoptera , Halictidae )
NGS / phylogénie / hétéroplasmie / Halictus smaragdulus / Wolbachia
Verwendung von Next Generation Sequencing zur Verbesserung des DNA Barcoding: Erfahrungen aus einer kleinen Studie an Wildbienen (Hymenoptera, Halictidae)
NGS / Phylogenie / Heteroplasmie / Halictus smaragdulus / Wolbachia
Electronic supplementary material
ESM 1.
(PPTX 132 kb)
Rights and permissions
About this article

Cite this article
Sonet, G., Pauly, A., Nagy, Z.T. et al. Using next-generation sequencing to improve DNA barcoding: lessons from a small-scale study of wild bee species (Hymenoptera, Halictidae). Apidologie 49, 671–685 (2018). https://doi.org/10.1007/s13592-018-0594-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-018-0594-y