
Abstract
Apis cerana Fabricius, the principle mainland species of Apis s.str. in Asia, remains an amalgamation of populations with considerable variation and regionally specialized morphotypes. In this study, populations of A. cerana were evaluated from the Himalayan region and areas in southwestern China as well as from neighboring Nepal based on a 97-bp-long fragment of mtDNA spanning the COI and COII genes. A total of 14 haplotypes were detected among the 58 sampled sequences, including 11 new haplotypes and 3 haplotypes previously reported from Chinese and Japanese populations of A. cerana. Analysis of these haplotypes and additional previously reported haplotypes from the Oriental region using neighbor-joining methods support a clustering of the Oriental haplotypes relative to southern India, but was unable to resolve with great support affinities within the branch. The network analysis revealed a mostly radiant genetic pattern, whereby most haplotypes were directly connected to one particular haplotype, Japan1. Our results indicate that the previously identified morphoclusters may reflect regional, ecological specialization among otherwise unrelated genetic lineages rather than a single, shared history.
Similar content being viewed by others
1 Introduction
Although honey bees (genus Apis L.) occur worldwide, much of their distribution is greatly influenced by humans owing to their importance in agriculture and honey production. The species living today are native to Europe, Africa, and southern Asia, but have been spread by human activity into the Americas, Australia, and most island ecosystems. While much of this movement represents the transfer of a single species, the Western Honey Bee (Apis mellifera L.) (Collet et al. 2006), its eastern counterpart, Apis cerana Fabricius, is equally subjected to significant population transfers. The ability of these species to be so amenable to movement is largely due to their considerable natural variability (Ruttner 1988; Baskaran 2011), and that permits populations to persist in habitats as varied as cold temperate to wet tropical and at elevations from sea level to high mountains within the Himalayas. It is therefore not surprising that A. cerana may be found from Far East Russia, Japan, and the Korean Peninsula, southward into the Philippines, Malaysia, and Indonesia, and west to easternmost Afghanistan and Pakistan, a vast area representing a plethora of specialized habitats (Oldroyd and Wongsiri 2006; Radloff et al. 2010, 2011; Hepburn and Radloff 2011). Since the 1980s, A. cerana has been moved to areas outside its natural range (namely New Guinea, the Solomon Islands, and Australia) (Anderson et al. 2010; Koetz 2013), and this has raised concerns regarding the competition between the invasive A. cerana and A. mellifera in Australia (Remnant et al. 2014), as well as the effect on the native fauna and flora (Koetz 2013). For example, introduced A. mellifera has been observed to impact native nectar-feeding bird populations in Australia (Paton, 1993).
Apis cerana is one of four species recognized in the nominate subgenus, Apis s.str., and the remaining species belong to the giant honey bees (subgenus Megapis) and the dwarf honey bees (subgenus Micrapis) (Engel 1999). There are also three extinct subgenera with a variety of species known from the Early Oligocene through Late Miocene, and that apparently form a grade relative to the extant groups (Engel 1998, 1999, 2006; Engel et al. 2009; Kotthoff et al. 2011, 2013). Within Apis s.str., A. mellifera is the earliest-branching species, representing the extant sister to the clade comprising A. cerana, Apis koschevnikovi Enderlein, and Apis nigrocincta Smith (Engel 1999; Engel and Schultz 1997; Radloff et al. 2010). Based on fossil evidence, the geographic origin of Apis s.l. was western Eurasia during the Early Oligocene (Kotthoff et al. 2013). The lineage of Apis s.str. apparently expanded its distribution through Africa and Asia during the Miocene, the former by the ancestor of A. mellifera and the latter by the common ancestor of A. cerana and its close relatives (Kotthoff et al. 2013). Once in Asia, the A. cerana-like populations were greatly influenced by Pliocene and Pleistocene climatic events which isolated groups that subsequently maintained their genetic isolation and are recognized as distinct species today, namely A. koschevnikovi and A. nigrocincta (Ruttner 1988; Radloff et al. 2011). Apis cerana, the principal mainland species of Apis s.str. in Asia, remains an amalgamation of populations with considerable variation and regionally specialized morphotypes (Tan et al. 2008; Radloff et al. 2011). Past authors had classified these highly variable populations into as many as 31 putative subspecies (Ruttner 1988; Engel 1999; Radloff et al. 2010), the validity of which was considered to be of little value given the significant degrees of overlap among these infraspecific units (Radloff et al. 2010). Nonetheless, there are distinctive patterns among the diverse A. cerana populations, and these may reflect local adaptation to specialized conditions, but this remains to be verified by extensive survey data (e.g., Arias and Sheppard 2005).
A recent, large-scale multivariate analysis of morphometric traits across A. cerana populations revealed six distinct morphoclusters, among them a “Himalayan cluster” consisting of bees of northern India, Tibet, and Nepal, and a “Northern cluster” comprising bees from most of China and areas as far west as northern Afghanistan and Pakistan (Radloff et al. 2010). The purpose of this study is to evaluate the regional-scale genetic structure of Eastern honey bee populations from across the Himalayan region, southwestern China, and Nepal using those mtDNA markers employed in previous studies (Cornuet et al. 1991; Garnery et al. 1992; Nikolenko and Poskryakov 2002; Tan et al. 2007). In addition, the data were used to detect whether or not there exists distinct genetic divisions within the region and to what extent there may be significant retrogression resulting from introductions by apiculturists.
2 Materials and methods
2.1 Sampling
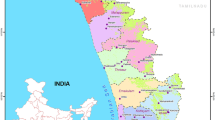
Worker bees representing 58 colonies of A. cerana were collected from local apiaries around the Himalayas and southwestern China, including five provinces in China and an area in Nepal (Figure 1). Specific locality information is provided in Table I. Bee samples were preserved in ethanol (90 %) and stored at −70 °C in the laboratory of the Eastern Bee Research Institute, Yunnan Agricultural University.

Locations of sampling sites across the Himalayan region.
2.2 DNA extraction and PCR amplification
Genomic DNA was extracted from the thorax of individual bees using the classical phenol-chloroform method (Smith and Hagen 1996). The mtDNA fragment used for this project spans the COI and COII genes and includes both coding and non-coding regions. PCR primers were designed using the software Oligo 6:
-
R: 5′–3′CTATACCTCGACGATACTCAG (21 bp) (Cytochrome Oxidase I)
-
F: 5′–3′TCAGGGTATTCATAGGATC (19 bp) (Cytochrome Oxidase II).
The positions of the primers correspond to nucleotides 3190–3210 and 3920–3947, respectively, of the complete mtDNA genome of A. cerana (NCBI accession no. GQ162109, Tan et al. 2011). We selected this intergenic region between COI and COII because it has been shown to be informative for infraspecific studies in both A. cerana (Baskaran 2011) and A. mellifera (Cornuet et al. 1991) owing to an apparent lack of purifying selection. PCR reactions were performed in a 25-μL reaction containing 18.4 μL sterile deionized water, 3.1 μL 10× PCR buffer (25 mmol Mg2+ concentration), 0.25 μL dNTPs (10 mM), 0.25 μL × 2 primers (20 mmol), 0.25 μL Taq DNA polymerase (2.5 U/μL; Takara), and 2.5 μL DNA template (70 ng/μL). The amplification reaction was pre-denatured at 95 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s each, annealing at 60.5 °C for 30 s, extension at 72 °C for 1 min, and a final extension at 72 °C applied for 10 min. PCR products were loaded onto 2 % agarose gels with ethidium bromide and examined under an ultraviolet trans-illuminator. PCR products were purified using a gel purification kit (Takara, China) and sequenced in both directions with corresponding PCR primers using an ABI 3730 sequencer system (Applied Biosystems, USA).
2.3 Sequence editing and analyses
One segment of the complete A. cerana mitochondrial genome (number GQ162109) was regarded as the target sequence (Tan et al. 2011). All sampled sequences were edited and checked using DNA Star 7.5 in order to generate the consensus sequences before they were subjected to multiple sequence alignment using Clustal X 2.0 (Larkin et al. 2007). Genetic characteristics such as haplotype diversity and total number of mutations were calculated using DnaSP version 5.10 (Rozas 2009).
For the analysis of haplotype relationships, the final dataset consisted of 22 haplotypes, including 11 haplotypes that were newly found in the present study, 3 previously reported haplotypes that were also recovered from our samples, and 8 additional haplotypes not found in our samples but retrieved from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) (Table II). An unrooted neighbor-joining (NJ) topology was reconstructed with the MEGA v6.06 software package (Tamura et al. 2013) and using the Kimura two-parameter model of nucleotide substitution (Kimura 1980). Support for branches in the NJ phenogram was estimated using 1000 bootstrap replicates. The three haplotypes from southern India were used as “outgroups” for rooting the optimal tree. Bayesian analysis was conducted using MrBayes 3.2 (Ronquist and Huelsenbeck 2003) and employed the best-fit nucleotide substitution model as implemented in jModelTest 0.1.1 (Posada 2009), with the Akaike information criterion set to a 100 % confidence interval (Akaike 1974). We considered two separate rounds of Markov chain Monte Carlo simulations of 4 chains and 1,000,000 generations each. Finally, the median-joining network of the haplotypes was constructed using Network v4.61 by Fluxus Technology (http://www.fluxus-engineering.com).
3 Results
3.1 Sequence variation and haplotype diversity
The total length of the studied sequences was 99 bp, with a total of 10 variable sites, or approximately 10 % of the full length. The 12 polymorphic sites included 10 substitutions, including 6 informative sites and 4 singletons, and 3 indels. The average content of T, A, C, and G was 39.1, 47.7, 8.3, and 4.9 %, respectively. The total content of A + T was 86.8 %, much higher than that of C + G, representing a significant bias similar to that across the non-coding region of the mtDNA genome of A. cerana. The nucleotide diversity was 0.00994 ± 0.00130 (Table III). When the 11 retrieved sequences were included, we found a total of 23 variable sites, which were 19 substitutions that including 6 informative sites, 13 singletons, and 4 indels (Figure 2).

Aligned haplotype sequences showing polymorphic sites. The haplotypes consisted of 11 new haplotypes detected in this study and 11 known haplotypes retrieved from GenBank, of which three were also found in the colonies sampled here.
3.2 Haplotype analyses
A total of 22 haplotypes were found among the 58 sampled sequences and the 11 retrieved sequences previously reported from Indian, Japanese, and Chinese populations of A. cerana. Among the 22 haplotypes, a total of 14 haplotypes were detected in our sampled sequences, including 11 newly identified haplotypes and 3 previously reported (Japan1, China2, and China4). The haplotype Japan1 was the most common, represented by 21 sequences in the present study. These sequences were mainly sampled from Nepal and Yunnan. Most haplotypes differed by only a single site mutation. The overall haplotype diversity of our samples was 0.705 ± 0.058 (Table III).
Putative relationships among the haplotypes were mostly unresolved except at the most basal levels when employing the NJ method as implemented in MEGA 6.06 (Figure 3) and when non-robustly supported nodes were collapsed. A group consisting of all mainland populations, including the haplotypes found in our samples and those reported previously, was relatively strongly supported as indicated by bootstrap values (Figure 3). However, most subsets within this mainland group can easily be collapsed because of low bootstrap support (Figure 3), except for two pairs of haplotypes and another group of four (Figure 3). Analysis using the more exhaustive Bayesian method was unable to resolve any relationships among the haplotypes, reflective of extensive reticulation and lack of population differentiation across the region.

Genealogical relationship among included haplotypes reconstructed using the NJ method as implemented in Mega 6.06 (Tamura et al. 2013). The tree is rooted using the three haplotypes of southern India. Numbers over the branches are bootstrap values based on 1000 replicates, and branches with a bootstrap value lower than 8 % were collapsed.
The network analysis of haplotypes clustered the samples from Yunnan with those from Nepal, whereas the haplotypes from the southern (Yunnan, Nepal) and northern (Tibet) populations did not form distinct clusters, respectively. The most common haplotype, Japan1, appeared as a comparatively ancestral haplotype among the studied populations, and to which almost all other haplotypes were directly connected and formed a linear relationship through. Two haplotypes were each directly connected with two other haplotypes, resulting in some degree of reticulate relationship among the haplotypes (Figure 4).

Genetic relationship among the included haplotypes of Apis cerana Fabricius reconstructed using the median-joining network method as implemented in Network v4.61 by Fluxus Technology (http://www.fluxus-engineering.com). Note: different sizes of the symbols reflect frequencies of all sequences in different haplotypes. (China1: 1; China2: 7; China3: 1; China4: 3; China5: 1; Japan1: 22; Japan2: 1; Japan3: 1; India1: 1; India2: 1; India3: 1; Nepal1: 3; Nepal2: 1; Nepal3: 1; Nepal4: 1; Qinghai: 9; Sichuan1: 2; Sichuan2: 3; Sichuan3: 1; Tibet: 4; Yunnan1: 2; Yunnan2: 2).
4 Discussion
The present analyses did not recover a strong divide between southern and northern populations in the Himalayan region (i.e., ranging between Nepal and Tibet) when compared with neighboring areas in southwestern China. This indicates that there is significant gene flow between honey bee populations on the two sides of the Himalayan range. Given that A. cerana is younger than the Himalayan uplift (Engel 1998, 1999; Kotthoff et al. 2013), it is perhaps not surprising that populations have migrated across this region and are not the result of a more ancient vicariant event. Moreover, it is likely that regional beekeepers have moved and traded colonies for centuries across the region, thereby bringing back into genetic contact populations that may have gradually become isolated over larger expanses of geological time. On the other hand, the haplotypes from the mainland form a relatively robust group in relation to those haplotypes from southern India (Figure 3). This basal separation between the Indian and mainland haplotypes probably reflects the dramatic climatic differences in India south of the Himalayas and the decreased influence of Chinese apicultural practices, and thereby much apiary contact, towards the south of India.
Among the 22 haplotypes included in our analyses, including 11 new and 11 previously reported, Japan1 was the most common, which is consistent with an earlier study that examined genetic diversity in Yunnan (Yin and Ji 2012). Based on our network reconstruction, Japan1 is putatively an ancestral haplotype given that almost all other haplotypes are directly connected to it and form a linear relationship (Figure 4). The only exceptions are the three haplotypes from India, which connected to Japan1 through multiple, intermediate hypothetical haplotypes, indicating an early separation from the putative ancestral Japan1 (Figure 4), and is similarly reflected in their higher genetic distance from Japan1 (Figure 3).
The morphometric variation observed by earlier studies (Radloff et al. 2010) may reflect some degree of regional specialization, but this may not be a fair characterization of genetic integrity within those clusters. For example, the Himalayan cluster identified in their analyses encompassed only populations existing at high elevations across the Himalayan range (represented herein by our populations from Nepal). One explanation for the clustering of these populations would be genetic continuity and isolation from the neighboring areas in Tibet and China. The analysis of mtDNA conducted herein within the region failed to recover an identical pattern (Figure 3). It therefore seems more likely that these morphoclusters reflect convergent specialization within a similar climate and habitat, rather than a cohesive genetic entity, with bees entering the geographic area from a variety of source populations and habitats (Radloff et al. 2005). Similarly, morphometric features, such as color and body size, found in the different mountainous populations, may be directly correlated to the special type of habitat (Nikolenko and Poskryakov 2002). For example, bees occurring in colder areas in mountains all tend to develop a darker coloration and more robust body size so as to maintain their temperature and more quickly heat up their flight muscles after the torpor of colder nights (Ruttner 1988; Tan et al. 2003, 2006).
Naturally, the samples here are relatively small given the total range of A. cerana. Nonetheless, the results indicate that well-characterized morphoclusters in A. cerana may reflect regional specialization among unrelated genetic lineages rather than a single, shared history. It will be important to expand such studies with denser sampling across the region and ultimately over the entire distribution of the species. Only through such comprehensive sampling and integration of data might it be possible to identify further areas and traits of convergent specialization among these hypervariable bees.
References
Anderson, D., Annand, N., Lacey, M., Ete, S. (2010) Control of Asian honeybees in the Solomon Islands. Australian Government, Australian Centre for International Agricultural Research.
Arias, M., Sheppard, W.S. (2005) Phylogenetic relationships of honey bees (Hymenoptera:Apinae:Apini) inferred from nuclear and mitochondrial DNA sequence data. Mol. Phylogenet. Evol. 37(1), 25–35
Baskaran, M. (2011) Mitochondrial DNA variations in Apis cerana populations of Tamil Nadu and Karnataka, South India. Biotechnol. Bioinf. Bioeng. 1(2), 221–227
Collet, T., Ferreira, K., Arias, M., Soares, A., Lama, M.D. (2006) Genetic structure of Africanized honeybee populations (Apis mellifera L.) from Brazil and Uruguay viewed through mitochondrial DNA COI–COII patterns. Heredity 97, 329–335
Cornuet, J., Garnery, L., Vautrin, D. (1991) Phylogenetic relationships in the genus Apis inferred from mitochondrial DNA sequence data. Apidologie 22, 87–92
Engel, M.S. (1998) Fossil honey bees and evolution in the genus Apis (Hymenoptera: Apidae). Apidologie 29(3), 265–281
Engel, M.S. (1999) The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae; Apis). J. Hymenoptera Res. 8(2), 165–196
Engel, M.S. (2006) A giant honey bee from the middle Miocene of Japan (Hymenoptera: Apidae). Am. Museum Novitates 3504, 1–12
Engel, M.S., Schultz, T.R. (1997) Phylogeny and behavior in honey bees (Hymenoptera: Apidae). Ann. Entomol. Soc. Am. 90(1), 43–53
Engel, M.S., Hinojosa-Díaz, I.A., Rasnitsyn, A.P. (2009) A honey bee from the Miocene of Nevada and the biogeography of Apis (Hymenoptera: Apidae: Apini). Proc. Calif. Acad. Sci., Series 4 60(3), 23–38
Garnery, L., Cornuet, J., Solignac, M. (1992) Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1, 145–154
Hepburn, H.R., Radloff, S.E. (2011) Honeybees of Asia. Springer, Verlag Berlin Heidelberg
Kimura, M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16(2), 111–120
Koetz, A.H. (2013) Ecology, behaviour and control of Apis cerana with a focus on relevance to the Australian incursion. Insects 4(4), 558–592
Kotthoff, U., Wappler, T., Engel, M.S. (2011) Miocene honey bees from the Randeck Maar of southwestern Germany (Hymenoptera, Apidae). ZooKeys 96, 11–37
Kotthoff, U., Wappler, T., Engel, M.S. (2013) Greater past disparity and diversity hints at ancient migrations of European honey bee lineages into Africa and Asia. J. Biogeogr. 40(10), 1832–1838
Larkin, M.A., Blackshields, G., Brown, N., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21), 2947–2948
Nikolenko, A.G., Poskryakov, A.V. (2002) Polymorphism of locus COI-COII of mitochondrial DNA in the honeybee Apis mellifera L. from the southern Ural Region. Russian J Genet 38(4), 364–368
Oldroyd, B.P., Wongsiri, S. (2006) Asian honey bees: biology, conservation, and human interactions. Harvard University Press, Cambridge
Paton, D.C. (1993) Honeybees in the Australian environment. BioScience 43(2), 95–103
Radloff, S.E., Hepburn, C., Hepburn, H.R., Fuchs, S., Hadisoesilo, S., Tan, K., Engel, M.S., Kuznetsov, V. (2010) Population structure and classification of Apis cerana. Apidologie 41(6), 589–601
Radloff, S.E., Hepburn, H., Engel, M.S. (2011) The Asian species of Apis. Honeybees of Asia. Springer, pp. 1-22.
Radloff, S.E., Hepburn, H.R., Fuchs, S., Otis, G.W., Hadisoesilo, S., Hepburn, C., Ken, T. (2005) Multivariate morphometric analysis of the Apis cerana populations of oceanic Asia. Apidologie 36(3), 475–492
Remnant, E.J., Koetz, A., Tan, K., Hinson, E., Beekman, M., Oldroyd, B.P. (2014) Reproductive interference between honeybee species in artificial sympatry. Mol. Ecol. 23(5), 1096–1107
Ronquist, F., Huelsenbeck, J.P. (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574
Rozas, J. (2009) DNA sequence polymorphism analysis using DnaSP. Bioinformatics for DNA sequence analysis. Springer, pp. 337-350.
Ruttner, F. (1988) Biogeography and taxonomy of honeybees. Springer-Verlag.
Smith, D.R., Hagen, R.H. (1996) The biogeography of Apis cerana as revealed by mitochondrial DNA sequence data. J. Kansas Entomol. Soc Supplement 69(4), 294–310
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729
Tan, H.W., Liu, G.H., Dong, X., Lin, R.Q., Song, H.Q., Huang, S.Y., Yuan, Z.G., Zhao, G.H., Zhu, X.Q. (2011) The complete mitochondrial genome of the Asiatic cavity-nesting honeybee Apis cerana (Hymenoptera: Apidae). PLoS One 6(8), e23008
Tan, K., Fuchs, S., Koeniger, N., Ruiguang, Z. (2003) Morphological characterization of Apis cerana in the Yunnan Province of China. Apidologie 34(6), 553–561
Tan, K., Hepburn, H.R., Radloff, S.E., Fuchs, S., Fan, X., Zhang, L., Yang, M. (2008) Multivariate morphometric analysis of the Apis cerana of China. Apidologie 39(3), 343–353
Tan, K., Meixner, M.D., Fuchs, S., Zhang, X., He, S., Kandemir, I., Sheppard, W.S., Koeniger, N. (2006) Geographic distribution of the eastern honeybee, Apis cerana (Hymenoptera: Apidae), across ecological zones in China: morphological and molecular analyses. Syst. Biodiv. 4(4), 473–482
Tan, K., Warrit, N., Smith, D.R. (2007) Mitochondrial DNA diversity of Chinese Apis cerana. Apidologie 38(3), 238–246
Yin, L., Ji, T. (2012) Genetic diversity of the honeybee Apis cerana in Yunnan, China, based on mitochondrial DNA. Genet. Mol. Res GMR 12(2), 2002–2009
Acknowledgments
We wish to thank Dr. Uma Partap for help with sampling bees in Nepal. This work was supported by the China National Research Fund (31260585) and the Key Laboratory of Tropical Forest Ecology of the Chinese Academy of Science. The participation of M.S.E. was partially supported by U.S. National Science Foundation grant DBI-1057366 (to M.S.E.). This is a contribution of the Division of Entomology, University of Kansas Natural History Museum.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript editor: Marina Meixner
Diversité de l’haplotype et similarité génétique parmi les populations de l’abeille orientale de l’Himalaya dans le sud-ouest de la Chine et du Népal (Hymenoptera: Apidae)
Apis cerana / région himalayenne / structure de la population / biogéographie / différenciation génétique / ADNmt
Diversität der Haplotypen und genetische Verwandtschaft in Populationen der östlichen Honigbiene , Apis cerana (Hymenoptera: Apidae), aus dem Himalaya-Gebiet in Südwest-China
Apis cerana / Himalaya-Region / Populationsstruktur / Biogeographie / genetische Differenzierung / mitochondriale DNA
Ken Tan and Yufeng Qu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Tan, K., Qu, Y., Wang, Z. et al. Haplotype diversity and genetic similarity among populations of the Eastern honey bee from Himalaya-Southwest China and Nepal (Hymenoptera: Apidae). Apidologie 47, 197–205 (2016). https://doi.org/10.1007/s13592-015-0390-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-015-0390-x