
Abstract
Purpose
This study seeks to determine if growth hormone-releasing hormone (GHRH) antagonist, JMR-132, increases survival when given before whole body radiation.
Material and methods
C3H mice were divided into 14 groups. The first 7 groups were given whole body radiation alone in the increasing doses of 7, 7.5, 8, 8.5, 9, 10, and 11 Gy, respectively. The other 7 groups received JMR-132 (10 μg/day s.c.) for 3 weeks then received the whole body radiation in increasing doses of 7, 7.5, 8, 8.5, 9, 10, and 11 Gy, respectively. The experiment lasted 35 days. Hazard ratios for survival were calculated for the addition of the GHRH antagonist as compared to radiation alone at the different dose levels. RT-PCR was performed to identify potential target genes. CD31 immunohistochemical staining identified the differences between average vessel count.
Results
Mice pretreated with JMR-132 showed a longer survival at lower radiation doses, the hazard ratios were 0.34 and 0.59 when the drug was given with doses of 7.5 and 8 Gy, respectively. A statistically significant higher hazard ratio of 3.48 and 4.22 was seen in mice when GHRH antagonist was added to doses of 10 and 11 Gy, respectively. Organ specific upregulation of several target genes such as Akt2, ATM, and Trp53 was seen with the GHRH antagonist. In the animals pretreated with JMR-132 the small intestine and the kidney showed higher average vessel count.
Conclusion
Pretreatment with GHRH antagonist JMR-132 protects at lower doses of radiation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiation can lead to damage of normal tissue that is dependent on radiation dose and volume irradiated. Whole body radiation from nuclear accidents can lead to acute radiation syndromes, involving the central nervous system, hematopoietic system, and gastrointestinal tract [1–3]. Whole body radiation can also be given therapeutically before transplantation. Thus, there is an important role for radioprotectants both in the medical field as well as in national defense. Radiation protectors, mitigators, as well as agents that address side effects of radiation damage have been developed, each with its own limitations. Examples include WR2721, pentoxyphylline, and alpha-tocopherol. [2]. New approaches being developed include radioprotection through suppression of apoptosis [4]. However, the need remains for an optimal radiation protection agent.
Growth hormone-releasing hormone (GHRH), secreted by the hypothalamus, denotatively regulates the release of growth hormone (GH) from the anterior pituitary gland. GHRH also functions as a tumor growth factor, as evidenced by the inhibition of growth of multiple experimental human cancers by GHRH-antagonists [5–7] and by the suppression of proliferation of breast, prostate, and lung cancer cell lines after the knocking down of gene expression [8, 9].
Our initial radiation experiments have already demonstrated that the sequence of treatment with GHRH antagonists is an important determinant of radiation response. Thus, increased tumor growth inhibition was seen when GHRH antagonists were given after the radiation, while a radioprotective effect, evidenced by an increase in tumor growth as compared to radiation alone, occurred when the GHRH antagonists were given before the radiation [10, 11].
The current study was designed to further investigate the radioprotective effect of GHRH antagonists in mice when given before whole body radiation (XRT). In parallel, PCR arrays, immunohistochemistry, and immunofluorescent staining were used to identify the potential target mechanisms of the radioprotective effect.
Materials and methods
Peptides and reagents
Peptides were synthesized in our laboratory by solid phase method and purified by reverse-phase HPLC as described previously [12, 13]. The structure of JMR-132 are the following:
Non-coded amino acids and acyl groups used in the antagonist are abbreviated as follows: Abu, alpha-aminobutyric acid; Cpa, 4-chloro-Phe; Har, homoarginine; Nle, norleucine; Tyr(Me), O-methyltyrosine. For in vivo studies, GHRH antagonist was dissolved in 0.1 % DMSO in 10 % aqueous propylene glycol solution (vehicle solution).
Animals
Twelve-week-old male C3H mice were obtained from the Charles River Laboratories (Wilmington, MA). The animals were housed in laminar airflow, conventional polycarbonate cabinets under pathogen-free conditions with a 12-h light/12-h dark schedule, and were fed autoclaved standard chow and water ad libitum. Experiments and animal care were conducted in accordance with institutional guidelines and complied with National Institute of Health animal policy.
Total body radiation
In vivo studies were carried out using 12-week-old C3H mice. Mice were randomized into 14 groups (n = 10). Radiation alone was given to the mice in groups 1 through 7 in an increasing doses of 7, 7.5, 8, 8.5, 9, 10, and 11 Gy, respectively, and consisted of whole body radiation using a 6 MVx X-ray beam (Varian Medical Systems). Mice in groups 8 through 14 were pretreated with the GHRH-antagonist JMR-132 (10 μg/day s.c.) for 3 weeks then received the whole body radiation in increasing doses of 7, 7.5, 8, 8.5, 9, 10, and 11 Gy, respectively. After 5 weeks, at the end of the experiment, mice were sacrificed under anesthesia, and necropsy was performed. Specimens from the kidney, liver, and intestines were snap frozen and stored at −70 °C, or immersed in phosphate-buffered 10 % formalin (pH 7.4) and embedded in paraffin for histological analysis.
Mouse cancer pathway finder real time PCR array
Total RNA was extracted and DNAse treated using the Macherey-Nagel NucleoSpin kit (Macherey-Nagel, Germany) from representative liver, kidney, and small intestinal tissues (three samples from each organ). Quantitative mRNA expression analysis of 84 genes representative of the major biological pathways involved in transformation and tumorigenesis was performed with the Mouse Cancer PathwayFinder™ RT2 Profiler™ PCR Array (SABioscience Corporation, Frederick, MD). The yield and the quality of RNA samples, synthesis of cDNA, and real-time RT-PCT arrays were performed as described [14]. Real-time PCR reactions were performed in the iCycler iQ™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA). All genes represented by the array showed a single peak on the melting curve characteristic of the specific products. Data analysis of gene expression was performed as described [16]. Briefly, using Excel based PCR Array Data Analysis software provided by manufacturer: fold-changes in gene expression were calculated using the ΔΔCt method and five stably expressed housekeeping genes (B2M, HPRT1, RPL13A, GAPDH, and ACTB) were used for normalization of the results.
CD31 staining
We used CD31 immunohistochemistry to quantify microvascular density. Paraffin embedded sections were cut into 4-μm sections, mounted onto slides, dewaxed in xylene, rehydrated in alcohol, and then endogenous peroxidase activity was blocked with 10 % hydrogen peroxide in water for 5 min. Antigen retrieval was performed by microwave treatment of the sections in citrate buffer for 20 min. Blood vessel density was determined by immunohistochemistry, using antibody to CD31 (1:50 dilution, sc-1506, Santa Cruz Biotechnology, Santa Cruz, CA), and a biotinylated rabbit IgG secondary antibody (1:100, BA-5000, Vector Laboratories, Burlingame, CA). The same concentration of rabbit IgG (sc-2027 Santa Cruz Biotechnology, Santa Cruz, CA) instead of the specific primary antibody was used as negative control. After incubation in avidin-biotin peroxidase complex for 30 min, 3,3′-diaminobenzidine tetrahydrochloride was used as chromogen. Between steps, the slides were rinsed three times for 5 min each in PBS. Sections were then counterstained with hematoxylin, dehydrated, and mounted. The number of CD31 positive vessels from three random high-powered fields (×400) of each section from three different organ samples was analyzed for each group. Images were captured using a Nikon Microphot-FXA microscope and a Nikon Coolpix 4300 digital camera (Nikon, Melville, NY). Vessel numbers were then measured in order to calculate the average value.
Statistical analysis
Hazard ratios for survival were calculated for the addition of GHRH antagonists as compared to radiation alone at different dose levels [17, 18]. Two-tailed Student’s t test was used where appropriate, and significance was accepted at p < 0.05. Results are expressed as means±SEM.
Results
In vivo effect of the GHRH antagonist JMR-132 on survival time and hazard ratio
One hundred and forty C3H mice were treated with either radiation alone or with JMR-132 followed by radiation. The C3H mice treated with GHRH antagonist showed a longer survival at lower radiation doses and a decreased survival at the highest doses of radiation. The hazard ratio was 0.34 with survival times of 9.2 ± 0.663 days vs. 14.3 ± 2.31 days without and with GHRH antagonist pretreatment at 7.5 Gy. For 8 Gy, it was 0.59 with survival times of 12.6 ± 0.733 days vs. 17 ± 3.018 days. This suggests a protective effect at the lower doses (Table 1). Statistically significant higher hazard ratios of 3.48 and 4.22 with survival times of 9 ± 0.577 days vs. 6.8 ± 0.918 days and 7.4 ± 0.266 days vs. 6.1 ± 0.1 days were seen in mice when JMR−132 was given with higher radiation doses of 10 and 11 Gy respectively.
Mouse cancer pathway finder real time PCR array
The mouse cancer pathway finder PCR array used in our study provided a simple and sensitive tool for profiling the expression of 84 genes related to cell proliferation, apoptosis, cell cycle, angiogenesis, invasion, and metastasis. We identified important target molecules leading to modulation of radiation response by GHRH antagonists. Nine genes in vital organs of mice exhibited significant change, relative to control, in mRNA expression when pretreatment with GHRH antagonist was followed by radiation.
In the kidney, we found significant upregulation of angiopoietin 1 (Angpt1), fibroblast growth factor receptor 2 (Fgfr2), non-metastatic cells 4, protein expressed in (Nme4), retinoblastoma 1 (Rb1), and thymoma viral proto-oncogene 2 (Akt2), while S100 calcium binding protein A4 (S100a4) was downregulated (p < 0.05 for all, see Table 2). In the liver, ataxia telangiectasia mutated homolog (Atm), S100a4 and tumor necrosis factor receptor superfamily, member 10b (Tnfrs10b) were found to be upregulated. In the small intestines, Angpt1, Akt2, and transformation related protein 53 (Trp53) were similarly upregulated (p < 0.05 for all, Table 2).
CD31 staining
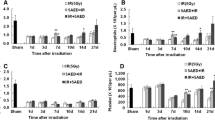
We used CD31 staining as a molecular method to detect neovascularization. CD-31, also known as platelet endothelial cell adhesion molecule-1 (PECAM-1), is a 130-kDa integral membrane protein and is expressed constitutively on the surface of adult and embryonic endothelial cells. Immunohistochemical studies showed remarkable differences between the control groups and the groups pretreated with JMR-132. The mean vessel counts per field on ×400 magnification in the control groups were 8.33 ± 0.88, 38.33 ± 1.76, and 5.66 ± 1.2 in the small intestine, liver, and kidney, respectively, whereas in the group pre-treated with JMR-132 the values were 10.67 ± 0.88, 23 ± 1.76, and 10.33 ± 1.2 in the small intestine, liver, and kidney, respectively (Fig. 1). We also found a correlation between the Angpt1 expression detected with RT-PCR and the average vessel count (Table 3).

Pretreatment with the GHRH antagonist JMR-132 increases the average vessel count in the small intestine (a) and in the kidney (c), but has no effect on the liver (b). Expression of CD31 appears as brown granules on representative slides from each organ (magnification, ×400; scale bar 20 μm)
Discussion
The finding that GHRH can function as a tumor growth factor [5–9, 12–16] increased interest in the development of GHRH antagonists. Antagonists of GHRH can exert both direct and indirect effects on tumors and other tissues. The indirect action is based on the suppression of growth hormone (GH) release from the pituitary, which leads to a reduction of hepatic insulin-like growth factor 1 (IGF-1) levels in the serum. The more important direct effect is produced by blockage of the autocrine/paracrine action of GHRH in tumors, as well as the autocrine/paracrine secretion inhibition of IGF-1 or IGF-2 from the tumors [7]. These result in growth inhibition demonstrated in multiple experimental human cancers [8, 9].
The sequence of treatment with GHRH antagonists and radiation was found to be a crucial determinant of response to radiation in prior experiments with prostate cancer (PC-3) and non-small cell lung carcinoma cell lines (H460) in nude mice. Increased tumor inhibition (radio-potentiation) occurred when GHRH antagonists were given after the radiation [10, 11]. Conversely, a tumor radioprotective effect was seen when GHRH antagonists were given before the radiation. Of note is that the 7 Gy dose level was an outlier and was repeated in another experiment with a similar GHRH-A (MZJ-7-138). The results confirmed a lower hazard ratio consistent with that ratio seen at the other similar dose levels [15]. The combined results of the previous and current experiment suggest a trend to an either radioprotectant/no effect with lower doses and a radiation sensitizing effect for higher doses. The exception is at 8.5 Gy, which will be confirmed in future experiments. Other contributing environmental factors in that particular group of mice that shared the same cage may have contributed to these results. This is the only unconfirmed dose point that is significantly different from others in the lower dose ranges of less than 9 Gy. However, until confirmatory studies are completed, the cut-off of 10 Gy will be referenced in our discussion of radiation sensitization.
It has already been demonstrated that induction of gonadal inhibition by treatment with LHRH agonists or antagonists can reduce the deleterious effects of chemotherapy and radiation on testicular and ovarian function [19–22]. With the exception of the 8.5 Gy findings discussed above, results from present and previous studies showed a radioprotective effect, with longer survivals, when GHRH antagonists were given before doses of less than 10 Gy of whole body radiation (XRT) in C3H mice. Although GHRH antagonists exert protective effects against damage inflicted by lower radiation doses, a radiosensitizing effect was seen at doses above 10 Gy, suggesting specific and different molecular changes leading to sensitization at the higher radiation doses.
Upregulation of target genes related to cell cycle, apoptosis, and recognition of damage was found to be organ specific and likely to contribute to the radioprotective effect described above. One of these genes, p53, is known to induce apoptotic death and cell cycle arrest [23, 24]. Furthermore, p53 is essential for solid organ protection against radiation damage and for maintenance of genomic integrity [23, 24]. The expression of wild-type p53 is already known to be upregulated in prostate cancer cells [25, 26] as well as hyperplastic benign prostate tissue treated with GHRH antagonists. Furthermore, intact p53 function is essential for solid organ protection against radiation damage and may be organ specific. This is especially seen at doses of radiation that can cause gastrointestinal radiation syndrome. At these doses, p53 may act as an inhibitor of mitotic death in the small intestine by inducing growth arrest [27]. p53-mediated growth arrest has a more significant impact on the extent of radiation-induced injury to the intestine than does p53-dependent apoptosis. Thus, p53 acts as a pro-survival rather than a pro-death factor in this situation. The opposite effect is seen in hematopoietic cells with gene mutations in p53, which become 2- to 3-fold more resistant to radiation due to loss of their ability to undergo late G1 arrest and to induce apoptosis [28]. It is important to note, however, that p53-mediated radioresistance may be more important in cells that depend on apoptosis, instead of necrosis, for cell death [29].
Although p53 was upregulated in all three sites (1.23-, 2.15-, and 69-fold in the liver, kidney, and small intestine, respectively), its elevation was most significant in the intestine. This is the first time that this significant overexpression of p53 in the intestine is reported in association with GHRH antagonist given before radiation. This p53 overexpression may partially explain some of the radioprotective effects seen in mice when GHRH antagonists were given before the radiation and may offer a radioprotective benefit to irradiated intestines.
Other related target genes studied include retinoblastoma 1 (Rb1), phosphatidylinositol 3′-kinase (PI3 kinase)/Akt signaling pathway genes, tumor necrosis factor receptor gene (TNF), and the ataxia telangiectasia mutated gene (ATM). Rb1 can suppress apoptosis with enhancement in cell survival after both ionizing radiation and ultraviolet radiation (UVR) [30, 31] and is upregulated in the kidney after treatment with GHRH-antagonist and may thus lead to its radioprotection. Downstream, the (PI3 kinase)/Akt signaling pathway is a major variable in radioresistance [32, 33]. Inhibition of either Akt1 or Akt2 results in enhanced radiosensitivity of some tumor cell lines [34]. In the current study, Akt2 was overexpressed in the small intestine and the kidney of mice treated with the GHRH antagonist, and may have contributed to the radioprotective effects when GHRH antagonists was given before radiation. It is also interesting that PI3 kinase signaling induced by ionizing radiation leads to the activation of Akt, specifically in endothelial cells [35, 36], and thus may have vascular endothelial protective, anti-apoptotic effects.
Ataxia telangiectasia mutated gene (ATM) induction is important in the pathway that ultimately leads to repair of radiation damage. It allows for DNA repair during p53 and checkpoint kinase 2 (CHK2) mediated G1/S or G2/M cell cycle block [37]. Conversely, cells defective in ATM or ATM-related protein (ATR) are hypersensitive to ionizing radiation [38, 39]. In this study, ATM was upregulated in the liver when GHRH antagonist were given before the radiation and may explain the radiation protective effect seen.
A more complex radiation interaction is seen with tumor necrosis factor-alpha (TNF-α). TNF-α not only protects against whole body radiation, but can also enhance the radiosensitivity of tumor cells in vitro [40]. These contradictory actions occur through different mechanisms. Activation of type I TNF receptors induces apoptosis through caspase-activation. This contrasts with its systemic radioprotective activity produced by induction of cytokines (IL-1 and IL-6), and activation of the survival-promoting nuclear factor kappa-B (NFκ-B), via the type II TNF receptor [41, 42]. The TNF receptor was also upregulated in the liver, approximately 10-fold.
The effect of GHRH antagonist on endothelial cells and the resulting vascular effects were also studied. Changes in intestinal crypt epithelial stem cells were historically, significantly thought to cause the lethal post-radiation gastrointestinal syndrome. However, recent evidence points to endothelial cells as the culprit. Paris et al. demonstrated that extensive apoptosis of endothelial cells was the primary lesion leading to stem cell dysfunction and initiation of intestinal radiation damage after abdominal radiation therapy in mouse models [43]. Endothelial cells can be protected against apoptosis through the action of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) [44, 45]. Furthermore, many in vitro studies suggest that angiopoietin-1 (Ang1) also has potential therapeutic applications in enhancing endothelial cell survival. For example, a potent Ang1 variant, COMP-Ang1 was studied in irradiated mice and was found to protect against radiation-induced apoptosis in microcapillary endothelial cells of the intestinal villi and leading to prolonged intestinal and systemic survival [46]. In the current study, angiopoietin is shown to be upregulated in both the small intestines and kidney. In addition, upregulation of fibroblast growth factor receptor in the kidney was also seen. These changes of vascular-associated gene mechanisms may have contributed to the observed radioprotective effect.
Staining for CD31 expression on the surface of endothelial cells has been used to demonstrate angiogenesis [47]. This method allows quantification of angiogenesis through immunohistochemical detection of CD31 [48]. In this study, the expression of angiogenesis measured by CD31 staining was consistent with the RT-PCR findings. An increase in the average vessel count was seen in the kidney and intestine, where RT-PCR also showed angiopoietin upregulation. In the liver, there was no angiopoietin upregulation on PCR and thus no change in the average vessel count.
Other factors may also play a role in the observed radiation protection of mice pretreated with GHRH antagonists. Additional possible explanations for the radioprotective effect may be related to the effect of GHRH antagonists on reactive oxygen species (ROS). ROS are produced by radiation and lead to radiation damage. Free radicals affect and regulate angiogenesis as well as radiotherapy response [49]. Previously, we examined the protein and lipid oxidative status of the LNCaP prostate cancer cell lines after treatment with GHRH and GHRH antagonists and found that cells exposed to GHRH antagonists generated fewer ROS than control cells [26]. Those results indicate that GHRH antagonists have strong antioxidant activity. Thus, the decrease in free radicals, induced by GHRH antagonists, may represent another mechanism for the radioprotective effect seen when GHRH antagonists were given before radiation.
These results confirm that GHRH antagonists given before radiation may have a radioprotective effect. However, there appears to be a radiation dose threshold for this radioprotective effect when given before whole body radiation as demonstrated, for the first time, in this study. The radioprotective effect may be explained by an upregulation of some potentially radioprotective targets. In addition, a vascular radioprotection mechanism may also significantly contribute to the radiation protection seen.
References
Stone HB, Moulder JE, Coleman CN, Ang KK, Anscher MS, Barcellos-Hoff MH, Dynan WS, Fike JR, Grdina DJ, Greenberger JS, Hauer-Jensen M, Hill RP, Kolesnick RN, Macvittie TJ, Marks C, McBride WH, Metting N, Pellmar T, Purucker M, Robbins ME, Schiestl RH, Seed TM, Tomaszewski JE, Travis EL, Wallner PE, Wolpert M, Zaharevitz D (2004) Models for evaluating agents intended for the prophylaxis, mitigation and treatment of radiation injuries. Report of an NCI Workshop, December 3–4, 2003. Radiat Res 162:711–718
Greenberger JS (2009) Radioprotection. In vivo 23:323–336
Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, Zhang L, Yu J (2008) PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2:517–518
Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, Kurnasov OV, Fort FL, Osterman AL, Didonato JA, Feinstein E, Gudkov AV (2008) An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 320:226–230
Schally AV, Varga JL (1999) Antagonistic analogs of growth hormone-releasing hormone: new potential antitumor agents. Trends Endocrinol Metab 10:383–391
Schally AV (2008) New approaches to the therapy of various tumors based on peptide analogues. Horm Metab Res 40:315–322
Schally AV, Varga JL, Engel JB (2008) Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab 4:33–43
Barabutis N, Schally AV (2008) Knocking down gene expression for growth hormone-releasing hormone inhibits proliferation of human cancer cell lines. Br J Cancer 98:1790–1796
Kiaris H, Schally AV, Kalofoutis A (2005) Extrapituitary effects of the growth hormone releasing hormone. Vitam Horm 70:1–24
Abdel-Wahab M, Schally AV, Szalontay L, Zarandi M, Varga JL, Pollack A, Rick FG (2009) The use of antagonists of growth hormone releasing hormone (GHRH) radioprotect tumors in vivo. Radiother Oncol 90(suppl 3):S113
Abdel-Wahab M, Schally AV, Szalontay L, Rick FG, Varga JL, Zarandi M, Pollack A (2009) Use of antagonists of growth hormone releasing hormone for protection during whole body radiation. Radiother Oncol 90(suppl 3):S107
Zarandi M, Horvath JE, Halmos G, Pinski J, Nagy A, Groot K, Rekasi Z, Schally AV (1994) Synthesis and biological activities of highly potent antagonists of growth hormone-releasing hormone. Proc Natl Acad Sci USA 91:12298–12302
Varga JL, Schally AV, Csernus VJ, Zarandi M, Halmos G, Groot K, Rekasi Z (1999) Synthesis and biological evaluation of antagonists of growth hormone-releasing hormone with high and protracted in vivo activities. Proc Natl Acad Sci USA 96:692–697
Rick F, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, Vidaurre I, Perez R, Halmos G, Szalontay L (2011) Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci 108(9):3755–3760
Abdel-Wahab M, Schally AV, Szalontay L, Rick FG, Varga JL, Shi Y-F, Zarandi M, Pollack A, Duncan R (2009) Radioprotection by antagonists of growth hormone releasing hormone given before whole body radiation is dependent on the radiation dose. Int J Radiat Oncol Biol Phys 75(3 Suppl):S552–S553
Rick FG, Schally AV, Szalontay L, Block NL, Szepeshazi K, Nadji M, Zarandi M, Hohla F, Buchholz S, Seitz S (2012) Antagonists of growth hormone-releasing hormone inhibit growth of androgen-independent prostate cancer through inactivation of ERK and Akt kinases. Proc Natl Acad Sci USA 109:1655–1660
Machin D, Cheung YB, Parmar MKB (2006) Survival analysis: a practical approach, 2nd edn. Wiley, Cambridge
Collett D (1994) Modeling survival data in medical research. Chapman & Hall, London
Schally AV, Paz-Bouza JI, Schlosser JV, Karashima T, Debeljuk L, Gandle B, Sampson M (1987) Protective effects of luteinizing hormone-releasing hormone against x-radiation-induced testicular damage in rats. Proc Natl Acad Sci USA 84:851–855
Jarrell J, YoungLai EV, McMahon A, Barr R, O’Connell G (1987) Effects of Ionizing Radiation and Pretreatment with [D-Leu6, des-Gly10] Luteinizing hormone-releasing hormone Ethylamide on developing rat ovarian follicles. Cancer Res 47:5005–5008
Ataya KM, McKenna JA, Weintraub AM, Clark MR, LeMaire WJ (1985) A luteinizing hormone-releasing hormone agonist for the prevention of chemotherapy-induced ovarian follicular loss in rats. Cancer Res 45:3651–3656
Huser M, Crha P, Ventruba P, Hudecek R, Zakova J, Smardova L, Kral Z, Jarkovsky J (2008) Prevention of ovarian function damage by a GnRH analogue during chemotherapy in Hodgkin lymphoma patients. Hum Reprod 23(4):863–868
Kastan MB (2007) Wild-type p53: tumors can’t stand it. Cell 128:837–840
Liu B, Chen Y (2008) St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med 44:1529–1535
Stangelberger A, Schally AV, Rick FG, Varga JL, Baker B, Zarandi M, Halmos G (2012) Inhibitory effects of antagonists of growth hormone releasing hormone on experimental prostate cancers are associated with upregulation of wild-type p53 and decrease in p21 and mutant p53 proteins. Prostate 72:555–565
Barabutis N, Schally AV (2008) Antioxidant activity of growth hormone-releasing hormone antagonists in LNCaP human prostate cancer line. Proc Natl Acad Sci USA 105(51):20470–20475
Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV (2004) Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23:3265–3271
O’Connor PM, Jackman J, Jondle D, Bhatia K, Magrath I, Kohn KW (1993) Role of the p53 tumor suppressor gene in cell cycle arrest and radiosensitivity of Burkitt’s lymphoma cell lines. Cancer Res 53:4776–4780
Pawlik TM, Keyomarsi K (2004) Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiation Oncology Biol Phys 59(4):928–942
Carlson CA, Ethier SP (2000) Lack of RB protein correlates with increased sensitivity to UV-radiation-induced apoptosis in human. Radiat Res 154(5):590–599
Haas-Kogan DA, Kogan SC, Levi D, Dazin P, Ang AT, Fung YK, Israel MA (1995) Inhibition of apoptosis by the retinoblastoma gene product. EMBO J 14(3):461–472
Testa JR, Tsichlis PN (2005) AKT signaling in normal and malignant cells. Oncogene 24:7391–7393
Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV (2005) The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 24:7482–7492
Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ (2005) Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res 65:7902–7910
Huber PE, Bischof M, Jenne J, Heiland S, Peschke P, Saffrich R, Gröne HJ, Debus J, Lipson KE, Abdollahi A (2005) Trimodal cancer treatment: beneficial effects of combined antiangiogenesis, radiation, and chemotherapy. Cancer Res 65:3643–3655
Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE (2002) Phosphatidylinositol 3-kinase/Akt signaling in the response of vascular endothelium to ionizing radiation. Cancer Res 62:4671–4677
Samuel T, Weber HO, Funk JO (2002) Linking DNA damage to cell cycle checkpoints. Cell Cycle 1:162–168
Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend S (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 17:159–169
Smith GC, Divecha N, Lakin NK, Jackson SP (1999) DNA-dependent protein kinase and related proteins. Biochem Soc Symp 64:91–104
Rosen EM, Fan S, Goldberg ID, Rockwell S (2000) Biological basis of radiation sensitivity part 2: cellular and molecular determinants of radiosensitivity. Oncology 14(5):741–757
Rothe M, Sarma V, Dixit VM, Goeddel DV (1995) TR4F2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science 269:1424–1427
Beg AA, Baltimore D (1996) An essential role for NF-kappa B in preventing TNF-alpha- induced cell death. Science 274:782–784
Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R (2001) Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 293:293–297
Haimovitz-Friedman A, Balaban N, McLoughlin M (1994) Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res 54:2591–2597
Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E (1995) Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1:1024–1028
Cho C-H, Kammerer RA, Lee HJ, Yasunaga K, Kim K-T, Choi H-H, Kim W, Kim SH (2004) Designed angiopoietin-1 variant, COMP-Ang1, protects against radiation-induced endothelial cell apoptosis. Proc Natl Acad Sci USA 101(15):5553–5558
DeLisser HM, Christofidou-Solomidou M, Strieter RM, Burdick MD, Robinson CS, Wexler RS, Kerr JS, Garlanda C, Merwin JR, Madri JA, Albelda SM (1997) Involvement of endothelial PECAM-1/CD31 in angiogenesis. Am J Pathol 151(3):671–677
Wang D, Stockard CR, Harkins L, Lott P, Salih C, Yuan K, Buchsbaum D, Hashim A, Zayzafoon M, Hardy RW, Hameed O, Grizzle W, Siegal GP (2008) Immunohistochemistry for the evaluation of angiogenesis in tumor xenografts. Biotech Histochem 83(3):179–189
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8:425–437
Acknowledgments
This study was supported by the Medical Research Service of the Veterans Affairs Department; the Division of Hematology/Oncology of the Miller Medical School, Departments of Pathology and Medicine, University of Miami; the South Florida Veterans Affairs Foundation for Research and Education (all A.V.S.); the L. Austin Weeks Endowment for Urologic Research (N.L.B.); the University of Miami Women’s Cancer Association (M.A.W.) and The Sylvester Cancer Center, University of Miami (M.A.W.).
Disclosure statement
The authors have nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdel-Wahab, M., Schally, A.V., Rick, F.G. et al. Antagonists of growth hormone releasing hormone (GHRH) given before whole body radiation lead to modulation of radiation response and organ-specific changes in the expression of angiogenesis. J Radiat Oncol 1, 389–396 (2012). https://doi.org/10.1007/s13566-012-0031-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13566-012-0031-1