
Abstract
Cancer cachexia is a debilitating consequence of disease progression, characterised by the significant weight loss through the catabolism of both skeletal muscle and adipose tissue, leading to a reduced mobility and muscle function, fatigue, impaired quality of life and ultimately death occurring with 25–30 % total body weight loss. Degradation of proteins and decreased protein synthesis contributes to catabolism of skeletal muscle, while the loss of adipose tissue results mainly from enhanced lipolysis. These mechanisms appear to be at least, in part, mediated by systemic inflammation. Exercise, by virtue of its anti-inflammatory effect, is shown to be effective at counteracting the muscle catabolism by increasing protein synthesis and reducing protein degradation, thus successfully improving muscle strength, physical function and quality of life in patients with non-cancer-related cachexia. Therefore, by implementing appropriate exercise interventions upon diagnosis and at various stages of treatment, it may be possible to reverse protein degradation, while increasing protein synthesis and lean body mass, thus counteracting the wasting seen in cachexia.
Similar content being viewed by others


1 Introduction
Cancer cachexia is a complex disorder characterised by a progressive weight loss, with catabolism of adipose tissue and skeletal muscle, affecting up to 50 % of cancer patients [1, 2]. Although cachexia may affect all cancer suffers, it is most commonly associated with those of the gastrointestinal tract and lungs. Patients with pancreatic or gastric cancer experience the highest frequency of weight loss, where patients can lose up to 30 % of their pre-illness weight. In contrast, patients with non-Hodgkin’s lymphoma, breast cancer and sarcomas show the lowest frequency of weight loss [2–4]. The exact reason for the frequent and profound severity in certain cancers is not known; however, it is thought that variations in tumour phenotype and host genotype may play a role in the development of cachexia [5].
The progressive weight loss seen in cachexia is an important prognostic factor for cancer survival, as greater weight loss is shown to be associated with shorter survival [6]. Cachexia accounts for 20 % cancer deaths occurring with 25–30 % total body weight loss. Even small amounts of weight loss can affect patients’ prognosis and treatment outcomes. For example, weight loss in breast and gastrointestinal cancer patients is associated with a decreased response to chemotherapy [7]. Lung cancer patients can experience extensive loss of both adipose tissue and skeletal muscle mass [8]. A 32 % weight loss from pre-illness stable weight was found to be associated with 85 % reduction in total body fat and 75 % loss of skeletal muscle. The marked loss of skeletal muscle seen in cachexia adversely impacts on the quality of life due to reduced mobility, fatigue and a decreased physical function [2] and ultimately a shorter life span. Loss of respiratory muscle function will lead to death from hypostatic pneumonia, and it is in fact respiratory failure that is responsible for death in 48 % of cancer suffers [9].
Although cachexia is most commonly associated with terminal stages of cancer, weight loss also commonly occurs in the early stages of the disease, with 17/20 patients with pancreatic cancer displaying a median weight loss of 14.2 % of pre-illness weight at the time of diagnosis resulting in a significantly reduced body mass index [8]. Studies also suggest that the rate and amount of weight loss are important factors in diagnosis and treatment of cancer, with poorer treatment outcomes being noted in patients with weight loss. This may be attributable to patients experiencing weight loss receiving significantly less chemotherapy and developing greater toxicity to treatment [7]. Therefore, a case can be made for trying to attenuate or prevent weight loss occurring upon diagnosis and tackling the effects of cachexia in the early stages of disease.
Several molecular mechanisms have been proposed to explain the pathways leading to the progressive muscle wasting seen in cachexia.
2 Molecular mechanisms of cancer cachexia
2.1 Adipose tissue breakdown
Loss of adipose tissue seen in cachexia is primarily due to an increase in lipolysis, as there is an increased turnover of glycerol and free fatty acids (FFA) in cachectic cancer patients compared with cancer patients with no weight loss or normal subjects [2, 10]. Cancer patients experiencing weight loss have been found to also have elevated levels of lipid mobilising factor (LMF), which is a tumour-induced catabolic factor that acts directly on the adipose tissue with the release of free fatty acids and glycerol [11, 12]. LMF binds with high affinity with β3-adrenegic receptor [11]. It is suggested that β-adrenergic activity plays an important role in the regulation of lipolysis, energy expenditure and triglyceride–fatty acid cycling in healthy populations [13]. In support of this, the administration of specific β-adrenergic receptor blockers and antagonists significantly reduces resting energy expenditure and fat mobilisation, in murine [11] and human studies [14], suggesting that these effects might, in part, be explained by increased β-adrenergic activity .
In addition, cancer cachexia also leads to an increased production of pro-inflammatory proteins that appear to mediate the wasting process via an increased local and systemic inflammation. [15].
2.2 Inflammation
Cachexia in humans appears to be associated with elevated levels of pro-inflammatory cytokines, tumour necrosis factor alpha (TNF-α), interferon-γ (IFN-γ) and interleukins (IL)-1 and IL-6 [16–18]. Being produced by both tumour and host, these pro-inflammatory cytokines have frequently been suggested as possible mediators in cachexia [4, 16, 19]. These are secreted by macrophages in reaction to infection, cellular damage or production of reactive oxygen species (ROS) and also by the tumour itself.
In addition to the loss of adipose tissue, cachexia also induces changes in lipid composition and structure, with increases in dimensions and alterations to ultrastructure of adipocytes being observed in tumour-bearing rats [20]. Further to this, adipocyte cell size and volume are positively correlated with the production of serum TNF-α [21, 22], a pro-inflammatory cytokine involved in many of the cachetic processes. Further over-expression of pro-inflammatory cytokines may, in turn, act in a synergistic manner, driving mechanisms responsible for the progressive wasting and furthering the cachetic state.
Experimental evidence also implicates TNF-α in the activation of protein turnover, leading to the loss of skeletal muscle [23–25]. However, in healthy young male subjects, 4 h infusion with recombinant human (rh) TNF-α did not affect skeletal muscle protein turnover [26]. In contrast, infusion with rhIL-6 for 3 h decreased muscle protein turnover by around 50 %, suppressing both synthesis and breakdown in healthy males. Despite that these findings may contradict the commonly held belief that TNF-α is the main mediator of cachexia, they are in accordance with the suggestion that pro-inflammatory cytokines act in a synergistic manner. It may also be plausible that certain human physiological pathways may even change as a result of long-term response, so it is still possible to reconcile these apparently contradictory findings and long-term exposure to excessive TNF-α may induce enhanced catabolism.
In addition to muscle protein breakdown, TNF-α has also been implicated in the stimulation of lipolysis [27] and the suppression of lipoprotein lipase (LPL). LPL is responsible for the hydrolysis of fatty acids from plasma lipoproteins, which are then transported into the adipocytes for synthesis of triacylglycerols [28].
Other cytokines have also frequently been implicated in the process of cachexia. Both IL-1 and 6 along with IFN-γ have been shown to inhibit expression of LPL mRNA, similar to TNF-α. Further to this, IL-1 and IFN-γ have been shown to directly stimulate lipolysis [2]. IL-6 levels have also been seen to correlate with the development of cachexia, with its neutralizing antibodies attenuating weight loss in cachexia [29]; similarly, the neutralizing antibodies of IFN-γ have the ability to slow weight loss. However, no significant relationship between levels and weight loss have been observed in cachectic cancer patients [30].
2.3 Muscle metabolism
Substantial loss of skeletal muscle is seen in cachexia. This is due mainly to an increase in whole body protein turn-over arising from an increased muscle protein breakdown. This is brought about by the degradation of muscle proteins via various proteolytic pathways; at the same time, a decrease in protein synthesis is observed [2, 4, 17]. However, the prominence and importance of synthesis and degradation vary between studies.
Decreases in protein synthesis may result from decreased plasma insulin concentrations and insulin sensitivity of the skeletal muscle, as metabolism of amino acids is in part altered by this decrease in insulin sensitivity [4, 17]. Early studies identified the role of insulin in promoting the movement of amino acids into striated muscle [31, 32], promoting protein synthesis and inhibiting degradation [33, 34]. Therefore, a decrease in insulin sensitivity of the skeletal muscle prevents the uptake of insulin and subsequently amino acids, suppressing protein synthesis.
TNF-α also appears to play an important role in the insulin resistance seen in cachexia, as insulin resistance and increased TNF-α levels are noted in several clinical syndromes with chronic inflammation [35, 36]. Insulin resistance seen in obese and diabetic patients is associated with increased TNF-α synthesis and secretion in the adipose cells [37–39]. Although the exact mechanisms are not fully known, TNF-α may be responsible for inhibiting the tyrosine phosphorylase activity of the insulin receptor leading to impaired insulin-stimulated glucose uptake [40]. Decreased protein synthesis may also result from decreased levels of protein translation, amino acid supply or the balance of amino acids required for protein synthesis [4]. Loss of physical activity in cachectic patients may also significantly affect the suppression of protein synthesis [4, 35]. Illness often results in a period of physical inactivity and bed rest. However, if prolonged with the absence of a stimulus afforded from physical activity, the metabolic homeostasis is compromised, resulting in the loss of lean muscle and physical function [41].
2.4 Protein catabolism
There are three major proteolytic pathways responsible for protein catabolism in skeletal muscle. The lysosomal system, which is mainly responsible for the degradation of extracellular proteins and cell receptors, has been implicated in inducing muscle wasting in the early stages of lung cancer. The increased mRNA levels of lysosomal protease cathepsin B presents an inverse relationship with body weight and fat-free mass (FFM) [42].
The cytosolic calcium-activated system which includes calpains I and II is mainly involved in tissue injury, necrosis and autolysis [43]. It has been suggested that the calcium/calpain pathway may act as an early, rate-limiting component of the catabolic process, releasing myofilaments from the sarcomere [44], later being catabolised by the third pathway. However, neither of these two pathways are capable of degrading myofibrillar proteins [45], and it is the myofibrillar proteins, actin and myosin, that are degraded in cachectic muscles [46, 47].
The third and most important pathway is the ubiquitin–proteasome pathway, which is dependent on ATP to dissemble and degrade muscle myofilaments [2, 43]. The bulk of intracellular proteins are degraded via the ubiquitin–proteasome pathway, in which proteins are marked for degradation via covalent attachments of chains to ubiquitin molecules. In gastric cancer patients experiencing an average of 6 % weight loss, the levels of ubiquitin mRNA were doubled, increasing to three times higher than control subjects in a similar study of patients suffering different types of cancers experiencing an average weight loss of 13 % [48, 49]. The ubiquitin chain is attached to the protein via a reaction sequence consisting of a series of three enzymes, E1 ubiquitin activation enzyme, E2 the ubiquitin carrier protein and E3 which recognises the ubiquitin protein ligase and catalyses the transfer of ubiquitin from E2 intermediate. Proteolysis is then catalysed by the 26S proteasome complex that degrades the marked proteins into small peptides [50–52]. Again pro-inflammatory cytokines, particularly TNF-α and IL-6, mediate this process.
The binding of TNF-α to its receptor site causes the activation of TNF receptor-associated factor, which is a ubiquitin ligase [15]. Similarly, both TNF-α and IL-6 induce the expression of E3α-II (a member of the E3 enzymes), which has been shown to be significantly induced at the onset and during progression of muscle wasting [53].
TNF-α is also thought to stimulate muscle catabolism via an NF–KappaB (Nf–kB)-dependent pathway, which increases muscle conjugation to muscle proteins [35, 54]. This occurs via a cascade of processes in which TNF-α binds to surface receptors, activating the Nf–kB pathway leading to the degradation of its inhibitory protein I-kBa. Nf–kB is then translocated to the monocyte nucleus where it alters gene expression, stimulating protein catabolism in the differentiated muscle cells [54, 55].
2.5 Oxidative stress
Oxidative stress due to increased production levels of ROS has also gained much attention for its possible involvement in cancer cachexia. Several possible mechanisms may increase oxidative stress within cancer patients. Firstly, loss of appetite, nausea and vomiting in cancer patients prevent the normal supply of nutrients, such as glucose, proteins and vitamins, eventually leading to the accumulation of ROS [56]. Secondly, non-specific activation of the immune system and excessive production of pro-inflammatory cytokines may in turn result in an in increase in ROS production [56, 57]. Proteins are one of the major targets of oxidative stress-derived effects on tissues [58]. In their study, Gomes-Marcondes and Tisdale [59] have shown that mild oxidative stress increases protein degradation through an increased expression of the major components of the aforementioned ubiquitin–proteasome pathway. This link between ROS and protein degradation has been noted in other studies utilising both in vivo and in vitro models [60, 61].
3 Exercise: potential mechanisms for preventing cachexia in cancer
Several treatment interventions aiming to reverse or restrict the progression of cachexia, include the use of pharmacological interventions with anticachectic agents and the targeting of inflammatory cytokines thought to mediate cachexia, mainly TNF-α [62, 63]; however, results remain equivocal. Anti-TNF treatment in patients with chronic inflammatory diseases experiencing cachexia reported no increase in muscle mass despite improvements in inflammatory markers, disease activity and physical function [64, 65]. Nutritional interventions, most notably, fish oil in combination with energy-dense nutritional supplements have been shown to increase lean body mass in cachectic cancer patients in some studies but not in others [66, 67].
Exercise could potentially be a promising intervention strategy for the prevention and treatment of cancer-related cachexia. With the ability to increase FFM, muscle strength and function, cardiovascular fitness and decrease fatigue, ultimately resulting in an increased quality of life, exercise may be an ideal strategy in helping to manage cancer-related cachexia [68, 69]. There is evidence to suggest that forms of exercise can be effective in slowing the progression of cachexia through several molecular mechanisms and anti-inflammatory effects [70–72]. In a recent review of cachexia in rheumatoid arthritis [36], it was concluded that physical exercise is the only therapeutic modality that has been shown to increase muscle mass, and should therefore form a major part of the management plan for early treatment in order to reduce cachexia and the burdens associated with the disease.
3.1 Exercise and inflammation
Exercise has the ability to reduce inflammation, with repeated exercise attenuating the cellular response to inflammatory stimuli and pro-inflammatory cytokines [73, 74].
Acute exercise is well known to induce an immune response [75, 76], with greatly enhanced production of both cytokines involved in an acute-phase inflammatory response and those that limit the inflammatory response [75, 77–81]. However, the fact that the classic pro-inflammatory cytokines, TNF-α and IL-1β, in general do not increase remarkably with exercise [70] indicates the cytokine cascade induced by physical activity differs from that induced by infections. The increase in circulating IL-6 in response to exercise has consistently been reported in the literature [78–84]. IL-6 is the first cytokine present in circulation during exercise, increasing in an exponential fashion up to 100-fold in relation to exercise intensity, duration and muscle mass recruited, declining in the post-exercise period [78, 83, 84]. Although often referred to as an inflammatory cytokine, IL-6 does not directly induce inflammation and has anti-inflammatory properties; therefore, it may be referred to as inflammatory-responsive [75]. Exercise is also associated with increased levels of well-known anti-inflammatory cytokine IL-10 and cytokine inhibitors, IL-1ra (IL-1 receptor antagonist), sTNF-r1 and sTNF-r2 (TNF receptors) that work as antagonists for inflammatory cytokines, blocking their ability to signal and mediate the aforementioned pathways [70, 77, 85]. Taken together, exercise evokes a primary increase in IL-6, which in turn, is followed by an increase in IL-ra and IL-10 [70, 86]. This anti inflammatory response to exercise may have the ability to reduce systemic inflammation and inflammatory cytokines, thus attenuating their role in mediating the wasting process in cachexia.
Evidence suggests that IL-6 mRNA is upregulated in skeletal muscle [79, 83, 87, 88], and the rate of transcription is significantly enhanced by exercise [89]. In addition to this, it has been shown that IL-6 protein is expressed in contracting muscle fibres and is released from skeletal muscle during exercise [78]. Strenuous exercise associated with eccentric muscle contractions resulting in muscle damage have repeatedly been associated with the immune response seen during exercise [79, 90, 91]. However, even moderate-intensity concentric exercise has a significant effect on muscle-derived IL-6. Performing 3 h of two-legged dynamic knee-extensor exercise at 50 % of their individual maximal power output resulted in only a moderate increase in heart rate but induced a 16-fold increase in IL-6 mRNA, 20-fold increase in plasma-IL-6 and a marked increase in IL-6 released from the working muscle in young healthy individuals [92]. Similarly, when the same model was applied to healthy untrained elderly individuals, greater amounts of IL-6 were released from the exercising muscles at the same relative intensity [93]. This suggests that cellular damage may not be responsible for the stimulation of cytokine production during exercise and that other metabolic and neuroendocrine factors may also contribute, as opposed to an immune response [94]. Further support for this comes from studies demonstrating that IL-6 mRNA in monocytes, the blood mononuclear cells responsible for the increase in plasma IL-6 during sepsis and other diseases, did not increase as a result of exercise [95, 96]. In addition, studies using flow cytometric techniques have demonstrated that the number, percentage and mean fluorescence intensity of monocytes staining positive for IL-6 do not change during cycling [97] and decrease during running [98]. These findings imply that the IL-6 response to exercise may not involve immune cell activation.
Studies suggest that IL-6 has anti-inflammatory effects, exerting inhibitory effects on pro-inflammatory cytokine (TNF-α and IL-1) production. IL-6 has a suppressive effect on both TNF-α and IL-1β production induced by the lipopolysaccharide [99]. Furthermore, elevated IL-6 infusion and exercise has been seen to attenuate endotoxin-induced increases in TNF-α in humans [74]. The fact that TNF-α levels are markedly elevated in anti-IL-6-treated mice [100, 101] adds further support that circulating IL-6 is involved in the regulation of TNF-α. Another anti-inflammatory IL-6 function is the stimulation of IL-1ra and IL-10 [86] anti-inflammatory cytokines, in addition to the release of soluble TNF-α receptors, sTNF-r1 and sTNF-r2 [77].
The appearance of circulating IL-10 and IL-1ra following exercise contributes to mediating the anti-inflammatory effect of exercise. IL-10 is the most important anti-inflammatory cytokine found within the human immune response [102], inhibiting the production of inflammatory cytokines IL-1α, IL-1β and TNF-α [102–104] through mRNA degradation of their corresponding genes [105]. Whilst IL-10 suppresses macrophage activities, IL-1ra inhibits signalling transduction through IL-1 receptor complex [106]. IL-1ra is part of the IL-1 family, binding to the IL-1 receptor sites at equal or greater affinity, blocking the action of IL-1α and IL-1β through competitive inhibition [102, 107]. In a similar fashion, TNF-soluble receptors (sTNF-r1 and sTNF-r2) bind to the molecule blocking its action [77, 108]. In addition to this, administration of sTNFr1 receptor has been seen to reduce the adverse effects of exaggerated TNF-α production observed in sepsis [108].
3.2 Exercise and inflammation in the adipose tissue
The progressive loss of adipose tissue seen with cachexia is predominantly due to an increase in lipolysis [2, 4] stimulated by TNF-α [27, 109]. Infusion of TNF-α increases whole-body lipolysis by 40 %, with a concomitant increase in FFA clearance [110]. Exercise has the ability to attenuate the action of TNF-α through increases in circulating anti-inflammatory cytokines as discussed above. In addition to this, the adipose tissue exhibits an altered inflammatory response to exercise. An 8-week endurance training programme at 60 % of VO2max, resulted in an increase of both IL-10 and TNF-α in the white adipose tissue of rats, with the principal effect being a large increase of IL-10 in the mesenteric depot, causing a change in the IL-10/TNF-α ratio, indicating an improved prognosis [72]. The IL-10/TNF-α ratio has been adopted as an indicator of an individual’s inflammatory status and disease-related morbidity, with lower values indicating a poor prognosis [111, 112]. Although both IL-10 and TNF-α increased, it is reasonable to speculate that the increase in TNF-α is related to the lipid metabolism by skeletal muscle during exercise, by inducing lipolysis [113]. Based on the evidence, it is plausible to think that the increase in IL-10 in response to endurance exercise seen in the study by Lira et al. [72] blocks the possible effects of TNF-α, including the stimulation of further lipolysis, with the increase in IL-10/TNF-α ratio adding further support to an anti-inflammatory environment.
3.3 Oxidative stress and exercise
ROS production may play an important role in the debilitating effects of cancer, directing muscle cells into a catabolic state [58, 59, 61]. On the other hand, exercise is well known to produce an anti-oxidative effect, through enhancing antioxidant enzyme activity [114–119]. Repeated exercise enhances the enzymes super-oxide dismutase (SOD) and glutathione peroxidase (GPx) activities in skeletal muscle [116, 117, 120], whereas mitochondrial Mn-SOD and catalase are induced in the lungs [121] and diaphragm [122, 123]. These antioxidative enzymes play an important role in protecting against the cell damage from ROS, with markers of damage being reduced following exercise [114].
Numerous non-enzymatic antioxidants exist in muscle cells, which offer protection from ROS. Glutathione (GSH) is one of the most important nonenzymatic antioxidants in muscle fibres. GSH is a tripeptide primarily synthesised in the liver and transported to tissues via circulation [124], with the higher concentrations being found in those tissues with high exposure to oxidants. Similarly, GSH concentration differs across the skeletal muscle fibres, with the highest concentrations being found in type I fibres [125]. Studies indicate that GSH increases in response to endurance exercise [125–127], which is most likely due to an increase in γ-glutamylcysteine, the rate-limiting enzyme for GSH biosynthesis [127, 128]. Exercise also appears to have a positive effect on other non-enzymatic antioxidants. Both α-lipoic acid and bilirubin, which appear to have strong anti-oxidant capabilities [129, 130], are increased during exercise [131–133]; however, their long-term exercise-induced effects still remain unknown [134, 135].
3.4 Exercise: molecular mechanisms of stimulating protein synthesis
3.4.1 GLUT-4, insulin sensitivity and exercise
Insulin sensitivity seen in cachectic patients has been implicated in the abnormal protein metabolism [38, 136]. The insulin resistance associated with cancer is not necessarily associated with malnutrition or stage of the disease, as normal glucose uptake is restored following tumour removal [137], suggesting that it may be tumour-induced. TNF-α has been proposed as a potential mediator in insulin resistance in cancer [37–39]; however, the aforementioned exercise-induced anti-inflammatory response may go some way to blocking the effects of TNF-α in mediating insulin sensitivity. In addition, exercise is also well known to enhance insulin sensitivity under non-inflammatory or cachectic conditions [138–142], indicating other mechanisms exist aside from the anti-inflammatory responses, to increase insulin sensitivity.
GLUT-4 are the glucose transport proteins responsible for mediating glucose transport into the skeletal muscle, with exercise enhancing their expression [143–145] and translocation [146–148]. Under normal conditions, these molecules reside in membrane vesicles inside the muscle cell; however, in response to insulin or muscle contraction, the GLUT-4 molecule translocates to the cell membrane where it inserts to increase glucose transport [149]. Several changes that occur during exercise appear to activate transcription factors to “turn on” the muscle GLUT-4 gene.
One of these changes is the decline in creatine phosphate (PCr) during exercise. An inverse correlation between GLUT-4 protein and high-energy phosphate levels has been noted in electronically stimulated muscles [150]. Furthermore, GLUT-4 protein was seen to be increased in rats with depleted PCr, again indicating a relationship [151]. AMP-activated protein kinase (AMPK) also stimulates GLUT-4 transcription. Activated during exercise, AMPK phosphorylates key target proteins that control flux through various metabolic pathways. Furthermore, it has been found to be dependent on two metabolic ratios AMP/ATP and creatine/PCr [152]. Activation of AMPK through injection of the adenosine analog 5-amino-imidazole-4-carboxamide ribonucleoside (AICAR) resulted in increased GLUT-4 expression fourfold in epitrochlearis muscles and 2.4-fold in gastrocnemius/plantaris muscles of rats [153]. Similarly, incubation of epitrochleris muscles with AICAR in vitro increased GLUT-4 expression [154].
Myocyte enhancer factor-2 (MEF2) is a transcription factor involved in intracellular signalling pathways controlling myogenesis and muscle hypertrophy [155], in addition to the exercise response and regulation of GLUT-4 [156–158]. MEF2 is activated through the calcium-dependent signalling pathway, in which calcineurin is activated by changes in intracellular calcium content (such as the increase seen with muscle contractions) [159, 160]. These changes enhance the number of GLUT-4 transporters that migrate to the cell membrane in response to insulin or muscle contraction, subsequently enhancing the transport of glucose and amino acids. This increase in transport of glucose and amino acids through enhanced insulin sensitivity would help to partially reverse the muscle wasting process through increasing substrate concentration.
3.4.2 Muscle metabolism, protein synthesis and resistance exercise
Resistance exercise is a potent stimulant for protein synthesis resulting in increases in muscle fibre cross-sectional area, particularly hypertrophy of myofibrillar proteins, myosin and actin [161, 162], stimulating both myofibrillar and mitochondrial protein synthesis by 67 % and 69 %, respectively [163]. Early studies suggested the possibility that muscle protein synthesis may be suppressed during acute bouts of exercise [164, 165]. However, the restriction of muscle protein synthesis that occurs during exercise is rapidly reversed following exercise recovery, a very consistent finding in the literature [166–171]. For example, muscle protein synthesis remained elevated for 72 h following 1 h of one-legged kicking exercise at 67 % of the individuals maximum workload [166] suggesting that, even moderate-intensity resistance exercise is effective at increasing protein synthesis.
The molecular regulation of muscle protein synthesis is complex and likely involves several interconnected cellular signalling pathways. The synthesis of protein is dependent on the transcription of DNA into mRNA, and the translation of mRNA into protein. A key regulator of this process appears to be insulin-like growth factor-1 (IGF-1) as induction of hypertrophy in adult skeletal muscle is accompanied by its increased expression [172–175]. Resistance exercise has a substantial effect on the expression of IGF-1 with increased IGF-1 mRNA concentrations being noted 48 h following exercise [176]. Furthermore, an increase of approximately 20 % has been noted in IGF-1 concentration during the first 13 weeks of a resistance training programme [177]. These reports suggest a relationship between stimulation of skeletal muscle cells through resistance training, IGF-1 expression and hypertrophy. The binding of IGF-1 to its receptor triggers the activation of several intracellular kinases, most notably dylinositol–3-kinase (PI3K), which in turn phosphorylates the membrane phospholipid phosphatidylinsitol–4,5-bis-phosphate, to phosphatidylinsitol–3,4,5-triphosphate, creating a lipid binding site on the cell membrane for a serine/threonine kinase known as Akt [178]. Akt is responsible for mediating cell growth and survival in a variety of tissues in response to IGF-1 [179–182] through its direct and indirect targets, glycogen synthase kinase 3 (GSK3), mammalian target of rapamycin (mTOR) and 70-kDa ribosomal protein S6 kinase (p70sk6), which are key regulators involved in protein translation and synthesis [183–186].
Resistance exercise leads to the activation of mammalian target of mTOR and its various kinases during immediate post-exercise recovery period [184, 187–189]. Furthermore, inhibition of this pathway through rapamycin, a specific inhibitor of mTOR, completely blocks muscle hypertrophy under growth conditions with several downstream components of the mTORC1 signalling pathway also being blunted or blocked by rapamycin [188], directly linking this pathway with muscle hypertrophy. Mounting evidence also implicates the role of extracellular-related kinase (ERK). ERK regulates the activity of several nuclear transcription factors in response to both diverse systemic stimuli, such as insulin and growth factors [190, 191] and local stressors, such as muscle contraction [49, 192]. ERK may also modulate mTOR activity through phosphorylating and inactivating tuberin–tuberous sclerosis complex (TSC2), a negative regulator of mTOR, inhibiting its ability to impair mTOR signalling [193]. Maximal eccentric muscle contractions activate 70-kDa ribosomal protein S6 kinase (p70sk6) [194], a downstream target of mTOR [192]. The degree of p70sk6 phosphorylation following a single bout of resistance exercise is strongly associated with long-term increase in muscle mass [195]. This was later reiterated in a study by Terzis et al. [196] who found that phosphorylation of p70sk6 in response to a single session of resistance exercise is strongly correlated to with an increase in FFM, squat 1-repetition maximum and hypertrophy of type II skeletal muscle fibres in response to a 14-week resistance training programme, suggesting that p70sk6 phosphorylation is involved in the signalling events leading to an increase in protein accretion in skeletal muscle following resistance exercise. Together, these findings highlight the potency of resistance exercise in stimulating protein synthesis, implying contractile activity may regulate muscle protein synthesis through the regulation and initiation of translation through several independent, but convergent pathways.
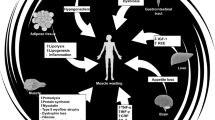
Figure 1 summarises the above-mentioned pathways and depicts the potential of aerobic and resistance exercises in attenuating the imbalance in protein catabolism and synthesis seen in cancer cachexia.

Key mechanisms responsible for the regulation of protein synthesis and catabolism in cancer cachexia and the influence of exercise. Key: IL = interleukin, TNFα = tumour necrosis factor alpha, sTNF-r = soluble TNF receptor, GLUT = glucose transporter, ROS = reactive oxygen species, SOD = super-oxide dismutase, GPx = glutathione peroxidase, GSH = glutathione, IGF-1 = insulin-like growth factor-1, AKt = protein kinase B, mTOR = mammalian target of rapamycin, p70 sk6 = 70-kDa ribosomal protein S6 kinase
3.5 Resistance training in cancer
Several studies have investigated the effectiveness of resistance training and its outcomes in cancer patients [197–204]. Of these studies, two investigated the effect of resistance training in breast cancer patients receiving adjuvant therapy [201, 202], in which women in the resistance training groups had a significant increase in lean body mass. In addition to this, the study by Courneya et al. [201] also reported increased chemotherapy completion rate. However, the fact that breast cancer is not typically associated with cachexia makes it difficult to generalise the results of these studies to cachectic patients.
Other studies observed the effects of resistance training in patients with prostate cancer receiving androgen deprivation therapy [198–200] and radiation therapy [205]. In these studies, resistance training was seen to prevent loss of muscle mass and strength seen in patients without resistance training [198, 199], increase serum growth hormone [200], reduce fatigue and improve quality of life [205]. Similar results were found in the Quist et al. [204] study, which investigated the application of high-intensity resistance training in cancer patients undergoing chemotherapy, reporting an average increase in muscle strength of 41.3 % and a significant increase in body weight of 1 %. Taken together, the aforementioned studies yield promising results for the application of resistance training in cancer patients for the maintenance of lean body mass, muscle strength and the reduction of fatigue associated with cancer treatment. However, because of the small number of existing clinical trials, it is difficult to make conclusions on the effects of resistance training in cachetic cancer patients.
3.6 Resistance training in muscle wasting disorders
Although little is known about the aforementioned intracellular signalling pathways that promote muscle protein synthesis in conditions of muscle wasting, evidence suggests that resistance training can increase muscle strength and lean body mass in muscle wasting disorders. The majority of research in this area has focused on aging populations experiencing sarcopenia, with resistance training being seen to increase muscle mass and strength in older adults and frail elderly individuals [206–209]. In addition to these adaptations, resistance training is also associated with increased functional performance of daily activities [210–212].
The potential of resistance exercise to increase muscle mass, strength and improve functional improvement has been extended to other muscle wasting disorders. A 12-week progressive resistance training programme increased muscle fibre size (type I and II) and improved muscle strength by 25–30 % in patients with renal disease. Similarly, Cheema et al. [213] reported significant improvements in muscle attenuation, strength, mid-thigh and mid-arm circumference, and body weight following a 12-week progressive resistance training programme during haemodialysis treatment. Resistance training has also been applied to try and counteract HIV-associated muscle wasting. Following 8 weeks of progressive resistance training, improvements in muscle strength of 60 % and lean body mass of 5 % were reported in men and women experiencing HIV-associated muscle wasting [214]. Similar results were found for women with HIV, following a 16-week supervised home exercise programme [215] and for men with HIV, seeing increases in muscle strength of 23–38 % on various exercises following a 16-week programme [216]. Although there is little evidence for the support of resistance training in cancer cachexia, the positive effects on lean muscle mass, strength and muscle function in other populations experiencing muscle wasting indicates that resistance training is an effective intervention for the attenuation of progressive muscle wasting.
4 Conclusions
The loss of both adipose tissue and skeletal muscle seen in cancer cachexia involves multiple independent, yet convergent molecular pathways, the majority of which appear to be, at least, partially mediated by chronic systemic inflammation and pro-inflammatory cytokines. TNF-α appears to work in a synergistic manner to enhance catabolism and has been implicated in the mechanisms of protein degradation and reduction in protein synthesis, through insulin resistance. Pro-inflammatory cytokines also appear to mediate the loss of adipose tissue seen in cachexia, through TNF-α-induced lipolysis. Exercise has been shown to have anti-inflammatory properties, through the upregulation of anti-inflammatory cytokines in skeletal muscle and adipose tissue, with the ability to block the actions of TNF-α and its ability to mediate the aforementioned mechanisms. In addition to its anti-inflammatory effects, exercise also enhances insulin sensitivity through interlinked molecular mechanisms, enhancing skeletal muscle metabolism. Resistance exercise has also been shown to be a potent stimulant for increasing protein synthesis and has been shown to reverse skeletal muscle wasting in other diseases, increasing muscle strength and lean body mass.
Therefore, regular exercise and physical activity may attenuate, and possibly reverse, the adverse effects of cancer cachexia through suppression of inflammatory burden that appears to drive the wasting process and enhancement of insulin sensitivity, protein synthesis and antioxidant enzymes. However, the literature surrounding the aforementioned mechanisms in cancer cachexia is scarce; future research must focus on the effects of exercise at attenuating wasting associated with cachexia and the molecular mechanisms involved.
References
Gordon JN, Green SR, Goggin PM. Cancer cachexia. QJM. 2005;98:779–88.
Tisdale MJ. Mechanisms of Cancer Cachexia. Physiol Rev. 2009;89:381–410.
Acharyya S, Guttridge DC. Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin Cancer Res. 2007;13:1356–61.
Tisdale MJ. Cachexia in cancer patients. Nat Rev Genet. 2002;2:862–71.
Monitto CL, Berkowitz D, Lee KM, Pin S, Li D, Breslow M, O’Malley B, Schiller M. Differential gene expression in a murine model of cancer cachexia. American Journal of Physiology - Endocrinology and Metabolism. 2001;281:E289–97.
Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass Jr HO, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am J Med. 1980;69:491–7.
Andreyev HJN, Norman AR, Oates J, Cunningham D. Why do patients with weight loss have worse outcomes when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer. 1998;34:503–9.
Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer with unresectable pancreatic cancer. Br J Cancer. 1997;75:106–9.
Windsor JA, Hill GL. Risk factors for postoperative pneumonia. The importance of protein depletion. Ann Surg. 1988;208:209–14.
Ryden M, Jocken J, van Harmelen V, Dicker A, Hoffstedt J, Wiren M, Blomqvist L, Mairal A, Langin D, Blaak E, Arner P. Comparative studies of the role of hormone-sensitive lipase and adipose triglyceride lipase in human fat cell lipolysis. Am J Endocrinol Metab. 2007;292:E1847–55.
Russell ST, Hirai K, Tisdale MJ. Role of b3-adrenergic receptors in the action of a tumour lipid mobilizing factor. Br J Cancer. 2001;86:424–8.
Russell ST, Tisdale MJ. Effect of a tumour-derived lipid-mobilising factor on glucose and lipid metabolism in vivo. Br J Cancer. 2002;87:580–4.
Klein S, Wolfe RR. Whole body lipolysis & triglyceride fatty acid cycling in cachectic patients with esophageal cancer. J Clin Invest. 1990;86:1403–8.
Hyltander A, Daneryd P, Sandström R, Körner U, Lundholm K. β-Adrenoceptor activity and resting energy metabolism in weight losing cancer patients. Eur J Cancer. 2000;36:330–4.
Lira FS, Rosa JC, Zanchi NE, Yamashita AS, Lopes RD, Lopes AC, Batista Jr ML, Seelaender M. Regulation of inflammation in the adipose tissue in cancer cachexia: effect of exercise. Cell Biochem Funct. 2009;27:71–5.
MacDonald N. Cancer cachexia and targeting chronic inflammation: a unified approach to cancer treatment and palliative supportive care. J Support Oncol. 2007;5:157–62.
Ardies CM. Exercise, cachexia, and cancer therapy: a molecular rationale. Nutr Cancer. 2002;42:143–57.
Argiles J, Lopez-Soriano FJ. The role of cytokines in cancer cachexia. Inc Med Res Rev. 1999;19:223–48.
Balkwill F, Mantovani A. Inflammation & cancer: back to Virchow? THE LANCET. 2001;357:539–45.
Bertevello PS, Seelaender MCL. Heterogeneous response of adipose tissue to cancer cachexia. Brazilian Journal of Medical and Biological Research. 2001;34:1161–7.
Winkler G, Kiss S, Keszthelyi L, Sapi Z, Ory I, Salamon F, Kovacs M, Vargha P, Szekeres O, Speer G, Karadi I, Sikter M, Kaszas E, Dworak O, Gero G, Cseh K. Expression of tumor necrosis factor (TNF)-alpha protein in the subcutaneous and visceral adipose tissue in correlation with adipocyte cell volume, serum TNF-alpha, soluble serum TNF-receptor-2 concentrations and C-peptide level. Eur J Endocrinol. 2003;149:129–35.
Morin CL, Gayles EC, Podolin DA, Wei Y, Xu M, Pagliassotti MJ. Adipose tissue-derived tumor necrosis factor activity correlates with fat cell size but not insulin action in aging rats. Endocrinology. 1998;139:4998–5005.
Llovera M, Garcia-Martinez C, Lopez-Soriano J, Agell N, Lopez-Soriano FJ, Garcia I, Argiles J. Protein turnover in skeletal muscle of tumour-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett. 1998;130:19–27.
Llovera M, Garcia-Martinez C, Lopez-Soriano J, Carbo N, Agell N, Lopez-Soriano FJ, Argiles J. Role of TNF receptor 1 in protein turnover during cancer cachexia using gene knockout mice. Mol Cell Endocrinol. 1998;98:S0303–72027.
Sherry BA, Gelin J, Fong Y, Marano M, Wei H, Cerami A, Lowry SF, Lundholm KG, Moldawer LL. Anticachectin/tumour necrosis factor alpha antibodies attenuate development of cachexia in tumour models. FASEB J. 1989;3:1956–62.
Petersen AM, Plomgaard P, Fischer CP, Ibfelt T, Pedersen BK, van Hall G. Acute moderate elevation of TNF-α does not affect systemic and skeletal muscle protein turnover in healthy humans. J Clin Endocrinol Metab. 2009;94:294–9.
Ryden M, Arvidsson E, Blomqvist L, Perbeck L, Dicker A, Arner P. Targets for TNF-alpha-induced lipolysis in human adipocytes. Biochem Biophys Res Commun. 2004;318:168–75.
Price SR, Olivecrona T, Pekala PH. Regulation of lipoprotein lipase synthesis in 3 T3–L1 adipocytes by cachectin. Further proof for identity with tumour necrosis factor. Biochem J. 1986;240:601–4.
Strassman G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest. 1992;89:1681–4.
Maltoni M, Fabbri L, Nanni O, Scarpi E, Pezzi L, Flamini E, Riccobon A, Derni S, Pallotti G, Amadori D. Serum levels of TNF-a and other cytokines do not correlate with weight loss and anorexia in cancer patients. Support Care Cancer. 1997;5:130–5.
Akedo H, Christensen HN. Nature of insulin action on amino acid uptake by the isolated diaphragm. J Biol Chem. 1962;237:118–22.
Manchester KL, Wool IG. Insulin and incorporation of amino acids into protein of muscle. 2. Accumulation and incorporation studies with the perfused rat heart. Biochem J. 1963;89:202–9.
Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest. 1987;80:1–6.
Fulks RM, Li JB, Goldberg AL. Effects of insulin, glucose, and amino acids on protein turnover in rat diaphragm. J Biol Chem. 1975;250:290–8.
Metsios GS, Stavropoulos-Kalinoglou A, Koutedakis Y, Kitas GD. Rheumatoid cachexia: causes, significance and possible interventions. Hospital Chronicles. 2006;1:20–6.
Summers GD, Metsios GS, Stavropoulos-Kalinoglou A, Kitas GD. Rheumatoid cachexia and cardiovascular disease. Nat Rev Rheumatol. 2010;6:445–51.
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–15.
Noguchi Y, Yoshikawa T, Marat D, Doi C, Makino T, Fukuzawa K, Tsuburaya A, Satoh S, Ito T, Mitsuse S. Insulin resistance in cancer patients is associated with enhanced tumor necrosis factor-alpha expression in skeletal muscle. Biochim Biophys Res Comm. 1998;253:887–92.
Nilsson J, Jovinge S, Niemann A, Reneland R, Lithell H. Relation between plasma tumour necrosis factor alpha and insulin sensitivity in elderly men with non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1998;18:1199–202.
Del Aguila LF, Claffey KP, Kirwan JP. TNF-a impairs insulin signaling and insulin stimulation of glucose uptake in c2c12 muscle cells. Am J Physiol Endocrinol Metab. 1999;276:E849–55.
Paddon-Jones D, Sheffield-Moore M, Cree MG, Hewlings SJ, Aarsland A, Wolfe RR, Ferrando AA. Atrophy and impaired muscle protein synthesis during prolonged inactivity and stress. J Clin Endocrinol Metab. 2006;91:4836–41.
Jagoe RT, Redfern CPF, Roberts RG, Gibson GJ, Goodship THJ. Skeletal muscle mRNA levels for cathepsin B, but not components of the ubiquitin-proteasome pathway, are increased in patients with lung cancer referred for thoracotomy. Clin Sci. 2002;102:353–61.
Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. American Society for Nutritional Sciences. J Nutr. 1999;129:227S–37.
Hasselgren P-O, Fischer JE. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg. 2001;233:9–17.
Lowell BB, Ruderman NB, Goodman MN. Evidence that lysosomes are not involved in the degradation of myofibrillar proteins in rat skeletal muscle. Biochem J. 1986;234:237–40.
Hasselgren P-O, James JH, Benson DW, Hall-Angerås M, Angerås U, Hiyama DT, Li S, Fischer JE. Total and myofibrillar protein breakdown in different types of rat skeletal muscle: effects of sepsis and regulation by insulin. Metabolism. 1989;38:634–40.
Long CL, Birkhahn RH, Geiger JW, Betts JE, Schiller WR, Blakemore WS. Urinary excretion of 3-methylhistidine: an assessment of muscle protein catabolism in adult normal subjects and during malnutrition, sepsis, and skeletal trauma. Metabolism. 1981;30:765–76.
Bossola M, Muscaritoli M, Costelli P, Bellantone R, Pacelli F, Busquets S, Argilès J, Lopez-Soriano LJ, Civello IM, Baccino FM, Fanelli FR, Doglietto GB. Increased muscle ubiquitin mRNA levels in gastric cancer patients. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1518–23.
Williamson D, Gallagher P, Harber M, Hollon C, Trappe S. Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. J Physiol. 2003;547:977–87.
Tisdale MJ. The ubiquitin–proteasome pathway as a therapeutic target for muscle wasting. J Support Oncol. 2004;3:209–17.
Lecker SH, Solomon V, Price SR, Tae KY, Mitch WE, Goldberg AL. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest. 1999;104:1411–20.
Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin–proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer. 2005;93:774–80.
Kwak KS, Zhou X, Solomon V, Baracos VE, Davis J, Bannon AW, Boyle WJ, Lacey DL, Han HQ. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res. 2004;64:8193–8.
Li P, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-alpha increases ubiquitin-conjugation activity in skeletal muscle by up-regulating UbcH2-E220k. FASEB J. 2003;17:1048–57.
Li P, Reid MB. NF-kB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1165–70.
Mantovani A, Madeddu C, Maccio A. Cancer-related anorexia cachexia syndrome and oxidative stress: an innovative approach beyond current treatment. Cancer Epidemiol Biomarkers Prev. 2004;13:1651–9.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16.
Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005;579:1646–52.
Gomes-Marcondes MCC, Tisdale MJ. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002;180:69–74.
Busquets S, Almendro V, Barreiro E, Figueras M, Argilés JM, López-Soriano FJ. Activation of UCPs gene expression in skeletal muscle can be independent on both circulating fatty acids and food intake: Involvement of ROS in a model of mouse cancer cachexia. FEBS Lett. 2005;579:717–22.
Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO. 1996;15:1753–65.
Gordon JN, Trebble TM, Ellis RD, Duncan HD, Johns T, Goggin PM. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut. 2005;54:540–5.
Goldberg RM, Loprinzi CL, Mailliard JA, O’Fallon JR, Krook JE, Ghosh C, Herstorff RD, Chong SF, Reuter NF. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. J Clin Oncol. 1995;13:2856–9.
Marcora SM, Chester KR, Mittal G, Lemmey AB, Maddison PJ. Randomized phase 2 trial of anti-tumor necrosis factor therapy for cachexia in patients with early rheumatoid arthritis. Am J Clin Nutr. 2006;84:1463–72.
Metsios GS, Stavropoulos-Kalinoglou A, Douglas KMJ, Koutedakis Y, Nevill AM, Panoulas VF, Kita M, Kitas GD. Blockade of tumour necrosis factor-α in rheumatoid arthritis: effects on components of rheumatoid cachexia. Rheumatology. 2007;46:1824–7.
Barber MD, Ross JA, Voss AC, Tisdale MJ, Fearon KC. The effect of oral nutritional supplement enriched with fish oil on weight loss in patients with pancreatic cancer. Br J Cancer. 1999;81:80–6.
Fearon KC, Barber MD, Moses AG, Ahmedzai SH, Taylor GS, Tisdale MJ, Murray GD. Double-blind, placebo-controlled, randomized study of eicosapentaenoic acid diester in patients with cancer cachexia. J Clin Oncol. 2006;24:3401–7.
Knols R, Aaronson NK, Uebelhart D, Fransen J, Aufdemkampe G. Physical exercise in cancer patients during and after medical treatment: a systematic review of randomized and controlled clinical trials. J Clin Oncol. 2005;23:3830–42.
Schneider CM, Hsieh CC, Sprod LK, Carter SD, Hayward R. Cancer treatment-induced alterations in muscular fitness and quality of life: the role of exercise training. Ann Oncol. 2007;18:1957–62.
Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98:1154–62.
de Lima C, Alves LE, Iagher F, Machado AF, Bonatto SJ, Kuczera D, de Souza CF, Pequito DC, Muritiba AL, Nunes EA, Fernandes LC. Anaerobic exercise reduces tumor growth, cancer cachexia and increases macrophage and lymphocyte response in Walker 256 tumor-bearing rats. Eur J Appl Physiol. 2008;104:957–64.
Lira FS, Rosa JC, Yamashita AS, Koyama CH, Batista Jr ML, Seelaender M. Endurance training induces depot-specific changes in IL-10/TNF-alpha ratio in rat adipose tissue. Cytokine. 2009;45:80–5.
Keller C, Keller P, Giralt M, Hidalgo J, Pedersen BK. Exercise normalises overexpression of TNF-alpha in knockout mice. Biochem Biophys Res Commun. 2004;321:179–82.
Starkie R, Ostrowski SR, Jauffred S, Febbraio MA, Klarlund B. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. FASEB J. 2003;17:884–6.
Pedersen BK, Hoffman-Goetz L. Exercise and the immune system: regulation, integration, and adaptation. Physiol Rev. 2000;80:1056–73.
Koch AJ. Immune response to exercise. Brazilian Journal of Boimorticity. 2010;4:92–103.
Ostrowski K, Rhode T, Asp S, Schjerling P, Pedersen BK. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol. 1999;51:287–91.
Steensberg A, Keller C, Starkie RL, Osada T, Febbraio MA, Pedersen BK. IL-6 and TNF-alpha expression in, and release from, contracting human skeletal muscle. Am J Physiol Endocrinol Metab. 2002;283:E1272–8.
Ostrowski K, Rhode T, Zacho M, Asp S, Pedersen BK. Evidence that interleukin–6 is produced in human skeletal muscle during prolonged running. J Physiol. 1998;508:949–53.
Leggate M, Nowell MA, Jones SA, Nimmo MA. The response of interleukin-6 and soluble interleukin-6 receptor isoforms following intermittent high intensity and continuous moderate intensity cycling. Cell Stress Chaperones. 2010;15:827–33.
Steensberg A, van Hall G, Osada T, Sacchetti M, Saltin B, Pedersen BK. Production of interleukin-6 in contracting human skeletal muscles can account for the exercise-induced increase in plasma interleukin-6. J Physiol. 2000;529(1):237–42.
Keller C, Steensberg A, Pilegaard H, Osada T, Saltin B, Pedersen BK, Neufer PD. Transcriptional activation of the IL-6 gene in human contracting skeletal muscle: influence of muscle glycogen content. FASEB J. 2001;15:2748–50.
Febbraio MA, Pedersen BK. Muscle-derived interleukin-6 mechanisms for activation and possible biological roles. FASEB J. 2002;16:1335–47.
Pedersen BK, Steensberg A, Schjerling P. Muscle-derived interleukin-6; possible biological effects. J Physiol. 2001;536(2):329–37.
Helmark IC, Mikkelsen UR, Borglum J, Rothe A, Petersen MC, Andersen O, Langberg H, Kjaer M. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: a randomized controlled trial. Arthritis Res Ther. 2010;12:R126.
Steensberg A, Fischer CP, Keller C, Moller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. 2003;285:E433–7.
Jonsdottir IH, Schjerling P, Ostrowski K, Asp S, Richter EA, Pedersen BK. Muscle contractions induce interleukin-6 mRNA production in rat skeletal muscles. Journal of Physiology. 2000;528:157–63.
Steensberg A, Febbraio MA, Osada T, Schjerling P, Van Hall G, Saltin B, Pedersen BK. Interleukin-6 production in contracting human skeletal muscle is influenced by pre-exercise muscle glycogen content. J Physiol. 2001;537(1):633–9.
Keller P, Keller C, Carey AL, Jauffred S, Fischer CP, Steensberg A, Pedersen BK. Interleukin-6 production by contracting human skeletal muscle: autocrine regulation by IL-6. Biochem Biophys Res Commun. 2003;310:550–4.
Rohde T, MacLean DA, Richter EA, Kiens B, Pedersen BK. Prolonged submaximal eccentric exercise is associated with increased levels of plasma IL-6. Am J Physiol Endocrinol Metab. 1997;273:E85–91.
Peake J, Nosaka K, Suzuki K. Characterization of inflammatory responses to eccentric exercise in humans. Exerc Immunol Rev. 2005;11:64–85.
Fischer CP, Hiscock NJ, Penkowa M, Basu S, Vessby B, Kallner A, Sjoberg LB, Pedersen BK. Supplementation with vitamins C and E inhibits the release of interleukin-6 from contracting human skeletal muscle. J Physiol. 2004;558:633–45.
Pedersen M, Steensberg A, Keller C, Osada T, Zacho M, Saltin B, Febbraio MA, Pedersen BK. Does the aging skeletal muscle maintain its endocrine function? Exerc Immunol Rev. 2004;10:42–55.
Moldoveanu AI, Shephard RJ, Shek PN. Exercise elevates plasma levels but not gene expression of IL-1β, IL-6, and TNF-α in blood mononuclear cells. J Appl Physiol. 2000;89:1499–504.
Ullum H, Haahr PM, Diamant M, Palmo J, Halkjaer-Kristensen J, Pedersen BK. Bicycle exercise enhances plasma IL-6 but does not change IL-1 alpha, IL-1 beta, IL-6, or TNF-alpha pre-mRNA in BMNC. J Appl Physiol. 1994;77:93–7.
Nehlsen-Cannarella SL, Fagoaga OR, Nieman DC, Henson DA, Butterworth DE, Schmitt RL, Bailey EM, Warren BJ, Utter A, Davis JM. Carbohydrate and the cytokine response to 2.5 h of running. J Appl Physiol. 1997;82:1662–7.
Starkie RL, Angus DJ, Rolland J, Hargreaves M, Febbraio MA. Effect of prolonged, submaximal exercise and carbohydrate ingestion on monocyte intracellular cytokine production in humans. J Physiol. 2000;528:647–55.
Starkie RL, Rolland J, Angus DJ, Anderson MJ, Febbraio MA. Circulating monocytes are not the source of elevations in plasma IL-6 and TNF-α levels after prolonged running. American Journal of Physiology - Cell Physiology. 2001;280:C769–74.
Schindler R, Mancilla J, Endres S, Ghorbani R, Clark SC, Dinarello CA. Correlations and Interactions in the production of interleukin-6 (IL-6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood. 1990;75:40–7.
Matthys P, Mitera T, Heremans H, van Damme J, Billiau A. Anti-gamma interferon and anti interleukin-6 antibodies affect staphylococcal enterotoxin B-induced weight loss, hypoglycemia, and cytokine release in d-galactosamine-sensitized and unsensitized mice. Infect Immun. 1995;63:1158–64.
Mizuhara H, O’Neil E, Seki N, Ogawa T, Kusunoki C, Otsuka K, Satoh S, Niwa M, Senoh H, Fujiwara H. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med. 1994;179:1529–37.
Opal SM. Anti-inflammatory cytokines. Chest. 2000;117:1162–72.
Opal SM, Wherry JC, Grint P. Interleukin-10: potential benefits and possible risks in clinical infectious diseases. Clin Infect Dis. 1998;27:1497–507.
Pretolani M. Interleukin-10: an anti-inflammatory cytokine with therapeutic potential. Clin Exp Allergy. 1999;29:1164–71.
Bogdan C, Paik J, Yoram V, Carl N. Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-beta and interleukin-10. THEJ OURNALO F BIOLOGICACLH EMISTRY. 1992;267:23301–8.
Dinarello CA. The role of the interleukin-1–receptor antagonist in blocking inflammation mediated by interleukin-1. N Engl J Med. 2000;343:732–4.
Svenson M, Hansen MB, Heegaard P, Abell K, Bendtzen K. Specific binding of interleukin 1 (IL-1)β and IL-1 receptor antagonist (IL-1ra) to human serum. High-affinity binding of IL-1ra to soluble IL-1 receptor type I. Cytokine. 1993;5:427–35.
Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci. 1992;89:4845–9.
Mathur N, Pedersen BK. Exercise as a mean to control low-grade systemic inflammation. Mediators Inflamm. 2008;2008:109502.
Plomgaard P, Fischer CP, Ibfelt T, Pedersen BK, van Hall G. Tumor necrosis factor-alpha modulates human in vivo lipolysis. J Clin Endocrinol Metab. 2008;93:543–9.
Kaur K, Sharma AK, Dhingra S, Singal PK. Interplay of TNF-alpha and IL-10 in regulating oxidative stress in isolated adult cardiac myocytes. J Mol Cell Cardiol. 2006;41:1023–30.
Leonidou L, Mouzaki A, Michalaki M, DeLastic AL, Kyriazopoulou V, Bassaris HP, Gogos CA. Cytokine production and hospital mortality in patients with sepsis-induced stress hyperglycemia. J Infect. 2007;55:340–6.
Nara M, Kanda T, Tsukui S, Inukai T, Younosuke S, Inoue S, Kobayashi I. Running exercise increases tumor necrosis factor-α secreting from mesenteric fat in insulin-resistant rats. Life Sci. 1999;65:237–44.
Ji LL. Antioxidants and oxidative stress in exercise. PSEBM. 1999;222:283–92.
Lawler JM, Powers SK, Visser T, Van Dijk H, Kordus MJ, Ji LL. Acute exercise and skeletal muscle antioxidant and metabolic enzymes: effects of fiber type and age. Am J Physiol. 1994;265:R1344–50.
Powers SK, Criswell D, Lawler JM, Martin AD, Herb RA, Dudley G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol. 1994;266:R375–80.
Berzosa C, Cebrian I, Fuentes-Broto L, Gomez-Trullen E, Piedrafita E, Martinez-Ballarin E, Lopez-Pingarron L, Reiter RJ, Garcia JJ. Acute exercise increases plasma total antioxidant status and antioxidant enzyme activities in untrained men. J Biomed Biotechnol. 2011;2011:540458.
Lambertucci RH, Levada-Pires AC, Rossoni LV, Curi R, Pithon-Curi TC. Effects of aerobic exercise training on antioxidant enzyme activities and mRNA levels in soleus muscle from young and aged rats. Mech Ageing Dev. 2007;128:267–75.
Miyazaki H, Oh-ishi S, Ookawara T, Kizaki T, Toshinai K, Ha S, Haga S, Ji LL, Ohno H. Strenuous endurance training in humans reduces oxidative stress following exhausting exercise. Eur J Appl Physiol. 2001;84:1–6.
Ji LL, Stratman FW, Lardy HA. Antioxidant enzyme systems in rat liver and skeletal muscle *1: influences of selenium deficiency, chronic training, and acute exercise. Arch Biochem Biophysics. 1988;263:150–60.
Duncan K., Harris S., and Ardies C.M. Running exercise may reduce risk for lung and liver cancer by inducing activity of antioxidant and phase II enzymes. Cancer Letters. 1997;116.
Oh-ishi S, Kizaki T, Ookawara T, Sakurai T, Izawa T, Nagata N, Ohno H. Endurance training improves the resistance of rat diaphragm to exercise-induced oxidative stress. AM J RESPIR CRIT CARE MED. 1997;156:1579–85.
Vincent HK, Powers SK, Stewart DJ, Demirel HA, Shanely AR, Naito H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur J Appl Physiol. 2000;81:67–74.
Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–76.
Leeuwenburgh C, Hollander J, Leichtweis S, Griffiths M, Gore M, Ji LL. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am J Physiol Regul Integr Comp Physiol. 1997;272:R363–9.
Ohkuwa T, Sato Y, Naoi M. Glutathione status and reactive oxygen generation in tissues of young and old exercised rats. Acta Physiologica Scandinavica. 1997;159:237–44.
Sen CK, Marin E, Kretzschmar M, Hanninen O. Skeletal muscle and liver glutathione homeostasis in response to training, exercise, and immobilization. J Appl Physiol. 1992;73:1265–72.
Kretzschmar M, Müller D. Aging, training and exercise. A review of effects on plasma glutathione and lipid peroxides. Sports Medicine (Auckland, NZ). 1993;15:196–209.
Packer L, Witt EH, Tritschler HJ. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19:227–50.
Barañano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci. 2002;99:16093–8.
Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci. 1987;84:5918–22.
Neuzil J, Stocker R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993;331:281–4.
Khanna S, Atalay M, Lodge JK, Laaksonen DE, Roy S, Hanninen O, Packer L, Sen CK. Skeletal muscle and liver lipoyllysine content in response to exercise, training and dietary α-lipoic acid supplementation. IUBMB Life. 1998;46:297–306.
Fallon KE, Sivyer G, Sivyer K, Dare A. The biochemistry of runners in a 1600 km ultramarathon. British Journal of Sports Medicine. 1999;33:264–9.
Chevion S, Moran DS, Heled Y, Shani Y, Regev G, Abbou B, Berenshtein E, Stadtman ER, Epstein Y. Plasma antioxidant status and cell injury after severe physical exercise. Proc Natl Acad Sci. 2003;100:5119–23.
Baracos VE. Regulation of skeletal-muscle–protein turnover in cancer-associated cachexia. Nutrition. 2000;16:1015–8.
Copeland GP, Leinster SJ, Daviis JC, Hipkin LJ. Insulin resistance in patients with colorectal cancer. Br J Surg. 1987;74:1031–5.
Wright DW, Hansen RI, Mondon CE, Reaven GM. Sucrose-induced insulin resistance in the rat modulation by exercise and diet. Am J Clin Nutr. 1983;38:879–83.
Henriksen EJ. Invited review: effects of acute exercise and exercise training on insulin resistance. J Appl Physiol. 2002;93:788–96.
Segerstrom AB, Glans F, Eriksson KF, Holmback AM, Groop L, Thorsson O, Wollmer P. Impact of exercise intensity and duration on insulin sensitivity in women with T2D. Eur J Intern Med. 2010;21:404–8.
Nassis GP, Papantakou K, Skenderi K, Triandafillopoulou M, Kavouras SA, Yannakoulia M, Chrousos GP, Sidossis LS. Aerobic exercise training improves insulin sensitivity without changes in body weight, body fat, adiponectin, and inflammatory markers in overweight and obese girls. Metabolism. 2005;54:1472–9.
Poehlman ET, Dvorak RV, DeNino WF, Brochu M, Ades PA. Effects of resistance training and endurance training on insulin sensitivity in nonobese, young women: a controlled randomized trial. J Clin Endocrinol Metab. 2000;85:2463–8.
Ren J, Semenkovich CF, Gulve EA, Gao J, Holloszy JO. Exercise induces rapid increases in GLUT4 expression, glucose transport capacity, and insulin-stimulated glycogen storage in muscle. J Biol Chem. 1994;269:14396–401.
Ivy JL. Muscle insulin resistance amended with exercise training: role of GLUT4 expression. MEDICINE & SCIENCE IN SPORTS & EXERCISE. 2004;36:1207–11.
Kraniou GN, Cameron-Smith D, Hargreaves M. Acute exercise and GLUT4 expression in human skeletal muscle: influence of exercise intensity. J Appl Physiol. 2006;101:934–7.
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5′AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1–5.
Kennedy JW, Hirshman MF, Gervino EV, Ocel JV. R. Armour Forse, Hoenig S.J., Aronson D., Goodyear L.J., and Horton E.S. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 1999;48:1–6.
Throell A, Hirshman MF, Nygren J, Jorfeldt L, Wojtaszewski JFP, Dufresne SD, Horton ES, Ljungqvist O, Goodyear LJ. Exercise and insulin cause GLUT-4 translocation in human skeletal muscle. Am J Physiol Endocrinol Metab. 1999;277:E733–41.
Dohm GL. Invited review: r egulation of skeletal muscle GLUT-4 expression by exercise. J Appl Physiol. 2002;93:782–7.
Yaspelkis 3rd BB, Castle AL, Farrar RP, Ivy JL. Contraction-induced intracellular signals and their relationship to muscle GLUT-4 concentration. Am J Physiol Endo. 1997;272:E118–W125.
Ren JM, Semenkovich CF, Holloszy JO. Adaptation of muscle to creatine depletion: effect on GLUT-4 glucose transporter expression. Am J Physiol Cell Physiol. 1993;264:C146–50.
Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch possible roles in type 2 diabetes. Am J Physiol Endocrinol Metab. 1999;277:E1–10.
Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–5.
Ojuka EO, Nolte LA, Holloszy JO. Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro. J Appl Physiol. 2000;88:1072–5.
Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–96.
Liu M, Olson AL, Edgington NP, Moye-Rowley W, Pessin JE. Myocyte enhancer factor 2 (MEF2) binding site is essential for C2C12 myotube-specific expression of the rat GLUT4 muscle-adipose facilitative glucose transporter. J Biol Chem. 1994;269:28514–21.
Thai MV, Guruswamy S, Cao KT, Pessin JE, Olson AL. Myocyte enhancer factor 2 (MEF2)-binding site is required for GLUT4 gene expression in transgenic mice. J Biol Chem. 1998;273:14285–92.
McGee SL, Hargreaves M. Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes. 2004;53:1208–14.
Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton J, Chin ER, Simard AR, Michel RN, Bassel-Duby R, Olson EN, Williams RS. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO. 2000;19:1963–73.
McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97:14400–5.
Hasten DL, Pak-Loduca J, Obert KA, Yarasheski KE. Resistance exercise acutely increases MHC and mixed muscle protein synthesis rates in 78–84 and 23–32 yr olds. American Journal of Physiology - Endocrinology And Metabolism. 2000;278:E620–6.
Balagopal P, Schimke JC, Ades P, Adey D, Nair KS. Age effect on transcript levels and synthesis rate of muscle MHC and response to resistance exercise. American Journal of Physiology - Endocrinology And Metabolism. 2001;280:E203–8.
Wilkinson SB, Phillips SM, Atherton PJ, Patel R, Yarasheski KE, Tarnopolsky MA, Rennie MJ. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol. 2008;586:3701–17.
Rennie MJ, Edwards RH, Krywawych S, Davies CT, Halliday D, Waterlow JC, Millward DJ. Effect of exercise on protein turnover in man. Clin Sci. 1981;61:627–39.
Rennie MJ, Edwards RH, Davies CT, Krywawych S, Halliday D, Waterlow JC, Millward DJ. Protein and amino acid turnover during and after exercise. Biochem Soc Trans. 1980;8:499–501.
Miller BF, Olesen JL, Hansen M, Dossing S, Crameri RM, Welling RJ, Langberg H, Flyvbjerg A, Kjaer M, Babraj JA, Smith K, Rennie MJ. Coordinated collagen and muscle protein synthesis in human patella tendon and quadriceps muscle after exercise. J Physiol. 2005;567:1021–33.
Biolo G, Maggi SP, Williams BD, Tipton KD, Wolfe RR. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am J Physiol. 1995;268:E514–20.
Kubica N, Bolster DR, Farrell PA, Kimball SR, Jefferson LS. Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin-dependent manner. J Biol Chem. 2005;280:7570–80.
Dreyer HC, Fujita S, Cadenas JG, Chinkes DL, Volpi E, Rasmussen BB. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J Physiol. 2006;576:613–24.
Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA, Smith K. Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol. 1991;73:1383–8.
Phillips SM, Tipton KD, Asle A, Wolf SE, Wolfe RR. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol. 1997;273:E99–107.
Adams GR, Haddad F. The relationships among IGF-1, DNA content, and protein accumulation during skeletal muscle hypertrophy. J Appl Physiol. 1996;81:2509–16.
Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol. 1998;84:1716–22.
Coleman ME, DeMayo F, Yin KC, Lee HM, Geskel R, Montgomeryl C, Schwartz R. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–16.
Goldspink DF, Garlick PJ, McNurlan MA. Protein turnover measured in vivo and in vitro in muscles undergoing compensatory growth and subsequent denervation atrophy. Biochem J. 1983;210:89–98.
Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, McLafferty CL, Urban RJ. Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab. 2001;280:E383–90.
Borst SE, De Hoyos DV, Garzarella L, Vincent K, Pollock BH, Lowenthal DT, Pollock ML. Effects of resistance training on insulin-like growth factor-I and IGF binding proteins. Med Sci Sports Exerc. 2000;33:648–53.
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Molecular Cell. 2004;14:395–403.
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7.
Datta SR, Brunet A, Greenberg ME. Cellular survival a play in three Akts. Genes Dev. 1999;13:2905–27.
Lawlor MA, Rotwein P. Insulin-like growth factor-mediated muscle cell survival central roles for Akt and cyclin-dependent kinase inhibitor p21. Molecular and Cell Biology. 2000;20:8983–95.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–88.
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeourt A, Lawrencet JC, Glass DJ, Yancopoulos GD. Akt-mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. NATURE CELL BIOLOGY. 2001;3:1014–9.
Nave BT, Ouwens MD, Withers DJ, Alessi DR, Shephard PR. Mammalian target of rapamycin is a direct target for protein kinase B identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–31.
Scott PH, Lawrence JC. Attenuation of mammalian target of rapamycin activity by increased cAMP in 3 T3-L1 adipocytes. J Biol Chem. 1998;273:34496–501.
Dreyer HC, Drummond MJ, Pennings B, Fujita S, Glynn EL, Chinkes DL, Dhanani S, Volpi E, Rasmussen BB. Leucine-enriched essential amino acid and carbohydrate ingestion following resistance exercise enhances mTOR signaling and protein synthesis in human muscle. Am J Physiol Endocrinol Metab. 2008;294:E392–400.
Drummond MJ, Fry CS, Glynn EL, Dreyer HC, Dhanani S, Timmerman KL, Volpi E, Rasmussen BB. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol. 2009;587:1535–46.
Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci USA. 2002;99:9213–8.
Tsakiridis T, Tsiani E, Lekas P, Bergman A, Cherepanov V, Whiteside C, Downey GP. Insulin, insulin-like growth factor-I, and platelet-derived growth factor activate extracellular signal-regulated kinase by distinct pathways in muscle cells. Biochem Biophys Res Commun. 2001;288:205–11.
Wojtaszewski JFP, Lynge J, Jakobsen AB, Goodyear LJ, Richter EA. Differential regulation of MAP kinase by contraction and insulin in skeletal muscle: metabolic implications. Am J Physiol Endocrinol Metab. 1999;277:E724–32.
Parkington JD, LeBrasseur NK, Siebert AP, Fielding RA. Contraction-mediated mTOR, p70S6k, and ERK1/2 phosphorylation in aged skeletal muscle. J Appl Physiol. 2004;97:243–8.
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93.
Eliasson J, Elfegoun T, Nilsson J, Kohnke R, Ekbolm B, Blomstrand E. Maximal lengthening contractions increase p70 S6 kinase phosphorylation in human skeletal muscle in the absence of nutritional supply. Am J Physiol Endocrinol Metab. 2006;291:E1197–205.
Baar K, Esser K. Phosphorylation of p70S6kcorrelates with increased skeletal muscle mass following resistance exercise. Am J Physiol. 1999;276:C120–7.
Terzis G, Georgiadis G, Stratakos G, Vogiatzis I, Kavouras S, Manta P, Mascher H, Blomstrand E. Resistance exercise-induced increase in muscle mass correlates with p70S6 kinase phosphorylation in human subjects. Eur J Appl Physiol. 2008;102:145–52.
Mustian KM, Peppone L, Darling TV, Palesh O, Heckler CE, Morrow GR. A 4-week home-based aerobic and resistance exercise program during radiation therapy a pilot randomized clinical trial. J Support Oncol. 2003;7:158–67.
Segal RJ, Reid RD, Courneya KS, Malone SC, Parliament MB, Scott CG, Venner PM, Quinney HA, Jones LW, Slovinec D’Angelo ME, Wells GA. Resistance exercise in men receiving androgen deprivation therapy for prostate cancer. J Clin Oncol. 2003;21:1653–9.
Galvao DA, Nosaka K, Taaffe DR, Spry N, Kristjanson LJ, McGuigan MR, Suzuki K, Yamaya K, Newton RU. Resistance training and reduction of treatment side effects in prostate cancer patients. Medicine & Science in Sports & Exercise. 2006;38:2045–52.
Galvao DA, Nosaka K, Taaffe DR, Peake J, Spry N, Suzuki K, Yamaya K, McGuigan MR, Kristjanson LJ, Newton RU. Endocrine and immune responses to resistance training in prostate cancer patients. Prostate Cancer Prostatic Dis. 2007;11:160–5.
Courneya KS, Segal RJ, Mackey JR, Gelmon K, Reid RD, Friedenreich CM, Ladha AB, Proulx C, Vallance JKH, Lane K, Yasui Y, McKenzie DC. Effects of aerobic and resistance exercise in breast cancer patients receiving adjuvant chemotherapy: a multicenter randomized controlled trial. J Clin Oncol. 2007;25:4396–404.
Schmitz KH, Ahmed RL, Hannan PJ, Yee D. Safety and efficacy of weight training in recent breast cancer survivors to alter body composition, insulin, and insulin-like growth factor axis proteins. Cancer Epidemiology Biomarkers & Prevention. 2005;14:1672–80.
Jones LW, Eves ND, Kraus WE, Potti A, Crawford J, Blumenthal JA, Peterson BL, Douglas PS. The lung cancer exercise training study: a randomized trial of aerobic training, resistance training, or both in postsurgical lung cancer patients: rationale and design. BMC Cancer. 2010;10:155–5.
Quist M, Rorth M, Zacho M, Andersen C, Moeller T, Midtgaard J, Adamsen L. High-intensity resistance and cardiovascular training improve physical capacity in cancer patients undergoing chemotherapy. Scandinavian Journal of Medicine & Science in Sports. 2006;16:349–57.
Segal RJ, Reid RD, Courneya KS, Sigal RJ, Kenny GP. Prud'Homme D.G., Malone S.C., Wells G.A., Scott C.G., and Slovinec D'Angelo M.E. Randomized controlled trial of resistance or aerobic exercise in men receiving radiation therapy for prostate cancer. J Clin Oncol. 2009;27:344–51.
Singh MF. DIng W., Manfredi T.J., Solares G.S., O’Neil E., Clements K.M., Ryan N.D., Kehayias J.J., Fielding R.A., and Evans W.J. Insulin-like growth factor I in skeletal muscle after weight-lifting exercise in frail elders. Am J Physiol Endocrinol Metab. 1999;277:E135–43.
Yarasheski KE, Pak-Loduca J, Hasten DL, Obert KA, Brown MB, Sinacore DR. Resistance exercise training increases mixed muscle protein synthesis rate in frail women and men≥ 76 yr old. Am J Physiol Endocrinol Metab. 1999;277:E118–25.
Rhodes EC, Martin AD, Taunton JE, Donnelly M, Warren J, Elliot J. Effects of one year of resistance training on the relation between muscular strength and bone density in elderly women. B J Sports Med. 2000;34:18–22.
Hakkinen K, Kraemer WJ, Newton RU, Alen M. Changes in electromyographic activity, muscle fibre and force production characteristics during heavy resistance power strength training in middle-aged and older men and women. Acta Physiol Scand. 2001;171:51–62.
Fiatarone MA, Marks EC, Ryan ND, Meredith CN, Lipsitz LA, Evans WJ. High-intensity strength training in nonagenarians. JAMA. 1990;263:3029–34.
Skelton DA, McLaughlin AW. Training functional ability in old age. Physiotherapy. 1996;82:159–67.
Macaluso A, Young A, Gibb KS, Rowe DA, De Vito G. Cycling as a novel approach to resistance training increases muscle strength, power, and selected functional abilities in healthy older women. J Appl Physiol. 2003;95:2544–53.
Cheema B, Abas H, Smith B, O’Sullivan A, Chan M, Patwardhan A, Kelly J, Gillin A, Pang G, Lloyd B, Singh MF. Progressive exercise for anabolism in kidney disease (PEAK): a randomized, controlled trial of resistance training during hemodialysis. J Am Soc Nephrol. 2007;18:1594–601.
Roubenoff R, Wilson IB. Effect of resistance training on self-reported physical functioning in HIV infection. Med Sci Sports Exerc. 2001;33:1811–7.
Dolan SE, Frontera W, Librizzi J, Ljungquist K, Juan S, Dorman R, Cole ME, Kanter JR, Grinspoon S. Effects of a supervised home-based aerobic and progressive resistance training regimen in women infected with human immunodeficiency virus. Arch Intern Med. 2006;166:1225–31.
Yarasheski KE, Tebas P, Stanerson B, Claxton S, Marin D, Bae K, Kennedy M, Tantisiriwat W, Powderly WG. Resistance exercise training reduces hypertriglyceridemia in HIV-infected men treated with antiviral therapy. J Appl Physiol. 2001;90:133–8.
Acknowledgments
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle 2010;1:7–8 (von Haehling S, Morley JE, Coats AJ and Anker SD).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Gould, D.W., Lahart, I., Carmichael, A.R. et al. Cancer cachexia prevention via physical exercise: molecular mechanisms. J Cachexia Sarcopenia Muscle 4, 111–124 (2013). https://doi.org/10.1007/s13539-012-0096-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-012-0096-0