
Abstract
This study reports a rapid and robust method for the differentiation of Asian and American ginseng samples based on differential ion mobility spectrometry-tandem mass spectrometry (DMS-MS/MS). Groups of bioactive ginsenoside/pseudo-ginsenoside isomers, including Rf/Rg1/F11, Rb2/Rb3/Rc, and Rd/Re, in the ginseng extracts were sequentially separated using DMS with stepwise changes in the gas modifier concentration prior to MS analysis. The identities of the spatially separated ginsenoside/pseudo-ginsenoside isomers were confirmed by their characteristic compensation voltages at specific modifier loading and MS/MS product ions. As expected, Asian ginseng samples contained some Rf and an insignificant amount of F11, whereas American ginseng samples had a high level of F11 but no Rf. The origin of the whole and sliced ginseng could further be confirmed using the quantitative ratios of three sets of ginsenoside markers, namely, Rg1/Re, Rb1/Rg1, and Rb2/Rc. Based on our results, new benchmark ratios of Rg1/Re < 0.15, Rb1/Rg1 > 2.15, and Rb2/Rc < 0.26 were proposed for American ginseng (as opposed to Asian ginseng).

Similar content being viewed by others


Introduction
Ginseng is a well-known natural product and has been used as a traditional Chinese medicine (TCM) for thousands of years in Asian countries. Many of the pharmacological effects that have been reported for ginseng, including anticancer and antioxidant properties and the modulation of cardiovascular conditions and immune function, have been attributed primarily to the presence of various ginsenosides as the major bioactive components [1]. Ginsenosides are a diverse group of steroidal saponins. Over 100 ginsenosides have been isolated from various species of ginseng [2]. Extensive phytochemical researchs on the structural characterization of ginsenosides and the differentiation of their isomers has been reported. The authentication of Panax ginseng (Asian ginseng), Panax quinquefolius (American ginseng), and ginseng products is typically conducted using chemical profiling [2, 3]. For example, ginsenoside Rf and pseudo-ginsenoside F11 are isomers, and are present solely in Asian ginseng and American ginseng, respectively [3]. Many analytical approaches have been used to identify ginsenosides, including liquid chromatography-mass spectrometry (LC-MS)-based methods [3, 4], tandem mass spectrometry (MS/MS) [5,6,7], thin-layer chromatography (TLC) [8], gas chromatography (GC) [9], near-infrared spectroscopy (NIRS) [10], and enzyme immunoassay [11]. According to previous reports, Asian and American ginseng can be differentiated using the presence/absence of Rf and F11; the Rb1/Rg1, Rg1/Re, and Rb2/Rc ratios; and the total panaxadiol/panaxatriol ratio [12]. The analytical challenge in their differentiation lies in the isomeric nature of various ginsenosides, such as Rf/F11/Rg1, Rd/Re, and Rb2/Rb3/Rc. One way to overcome this challenge is to use isomer-sensitive detection methods, such as NIRS and immunoassays [10, 11]. To utilize the ultrahigh sensitivity of mass spectrometry, a suitable separation technique, such as HPLC, must be applied for isomer separation prior to MS analysis.
Ion mobility spectrometry (IMS) is an ion separation technique that separates ions based on their mobilities in the gas phase, which depend on the size, charge number, and shape of the ions [13, 14]. Differential ion mobility spectrometry (DMS) (or field asymmetric waveform ion mobility spectrometry (FAIMS)) is a gas-phase ion separation technique that separates ions based on their mobility differences under high and low electric field conditions [15]. Further enhancement of the DMS separation efficiency is obtained by adding polar chemical modifiers to the buffer gas [16]. The change in the mobility behavior of the ions is considered to be caused by dynamic clustering and declustering processes involving the ions of interest and the buffer gas molecules [17]. Modifiers with specific chemical structures can result in distinctive selectivity enhancements [16, 18]. One major advantage of DMS is its ability to distinguish isobaric and isomeric compounds, including structural isomers [19], tautomers [20], and even stereoisomers [21]. Several studies have also demonstrated the applications of DMS for quantitative analysis [22,23,24].
This work reports a rapid and robust DMS-MS/MS method for the differentiation of Asian and American ginsengs. Our results demonstrated that the major ginsenoside/pseudo-ginsenoside isomers could be separated by DMS-MS/MS using different amounts of 1-propanol as the gas modifier. A rapid quantitative assay of the ratios of the three pairs of bioactive ginsenosides was developed for the differentiation of Asian and American ginsengs. Extracts from six commercially available ginseng samples were tested using this assay and were correctly authenticated on the basis of their origins.
Experimental Section
Reagents and Chemicals
Ginsenoside standards were purchased from WeiKeQi Biological Technology Co., Ltd. (Sichuan, China). Methanol and water were of LC-MS grade and were purchased from Sigma-Aldrich (St. Louis, USA). Acetonitrile was of LC-MS grade and was purchased from J. T. Baker (PA, USA). Anhydrous grade 1-propanol with 99.7% purity was purchased from Sigma-Aldrich (St. Louis, USA). Sodium acetate was obtained from Fluka (Neu-Ulmm, Germany). Six ginseng samples, comprising three American ginseng samples (USA whole ginseng, USA and Canadian sliced ginseng products) and three Asian ginseng samples (Korean whole ginseng, Chinese whole ginseng, and Chinese sliced ginseng product), were purchased via internet shops from different vendors in China between May and August 2018.
Sample Preparation
The ginseng samples were extracted, purified, and concentrated using a modified procedure from the literature [3] and then diluted before DMS analysis. Briefly, the ginseng sample was first homogenized. Of the powdered sample, 300 mg was extracted with 10 mL of chloroform (HPLC grade) under reflux for 3 h. The sample was then filtered, and the chloroform extract was discarded. The solid residue was extracted with 10 mL of methanol (HPLC grade) under the same conditions. The methanol solution was evaporated to dryness under vacuum. The solid was re-dissolved in 3 mL of deionized water (HPLC grade). The aqueous solution was applied to a SEP-PAK C18 cartridge (Waters Associates). The loaded cartridge was washed with 10 mL of water (HPLC grade) and then eluted with 10 mL of 50% acetonitrile (HPLC grade) in deionized water. The collected ginseng extracts were filtered through a 0.2-μm filter (Millipore) and were evaporated down to a volume of about 0.5 mL.
Instrumentation
A custom-built CaptiveSpray-DMS device was fitted onto the inlet capillary of a 9.4T SolariX FTICR mass spectrometer (Bruker Daltonics, Germany). Details of the CaptiveSpray-DMS device have been described previously [25]. Briefly, a standard CaptiveSpray source (Bruker Daltonics, Germany) was directly mounted at the entrance of a DMS device and maintained at a voltage of 3.0–3.5 kV. The DMS device comprises of a pair of planar electrodes of 20 mm wide and 80 mm long, spaced 1.4 mm apart. Samples were introduced into the CaptiveSpray source at a flow rate of 0.5 μL/min. CaptiveSpray gas was fed into the source to facilitate the collection of the sprayed ions into the co-axial inlet of the DMS chamber. Auxiliary gas was orthogonally inserted prior to the inlet of the DMS chamber and merged with the CaptiveSpray gas flow. The gas flow across the DMS chamber was controlled by the sum of the CaptiveSpray and auxiliary gas flows over a wide flow rate range without affecting the vacuum conditions of the inlet region of the mass spectrometer. The chemical modifier was delivered by a syringe pump and was added to a porous polyethylene column packed inside a T-shaped gas mixer. A continuous flow of nitrogen gas was used to evaporate the chemical modifier, which was split into the CaptiveSpray and the auxiliary gas. The modifier concentration was determined by the injection flow rate of the modifier solution and the flow rate of the nitrogen gas. As reflected from the shift of the compensation voltage (CoV) position of the ginsenoside standards, the modifier composition in the DMS carrier gas became steady within 2 min after changing the injection rate of the modifier solution. The inlet capillary high voltage (HV) was set to 0 V. Nitrogen gas was heated to 200–220 °C and was used as the drying gas at a flow rate of 1.3 L/min. In all the collision-induced dissociation experiments, argon was used as the collision gas. The precursor ions were isolated by a front-end quadrupole with a 5–10-Th window. The ion accumulation time in the hexapole was 0.1–0.6 s for the MS survey scan and 0.3–2.0 s for the MS/MS scan. Mass spectra were acquired with ftmsControl version 2.1 (Bruker Daltonics, Germany) in the broadband mode over a range of m/z between 150 and 1500. For MS/MS analysis using collision-induced dissociation, the following SRM ion transitions were selected: m/z 1131.5922 → 365.1054 for Rb1, m/z 1101.5816 → 789.4745 for Rb2, Rb3, and Rc, m/z 969.5368 → 789.4745 for Rd and Re, m/z 823.4807 → 365.1054 for Rf, m/z 823.4807 → 643.4178 for Rg1, and m/z 823.4807 → 497.3599 for F11. The collision energy was optimized in the range of 10–30 eV. The pulse program of the data-dependent acquisition was modified so as to synchronize the MS survey scan and the MS/MS scans with the external CoV scanning of DMS. The nomenclature for carbohydrate fragmentation used in this paper is based on the work of Domon and Costello [26]. The data were processed using Data Analysis 4.4 (Bruker Daltonics, Germany) and Origin Pro 8 (Origin Lab, Northampton, MA).
DMS Optimization and Quantitative DMS-MS/MS Analysis
1.0 μM solutions of each ginsenoside standard in methanol/water (50:50 v/v + 0.1% sodium acetate) were used in the DMS-MS experiments. The compensation voltage (CoV) was scanned over the range of − 50 to + 100 V while keeping the DMS dispersion field constant at 127 Td. 1-Propanol was used as the gas modifier for the DMS separation of isomeric ginsenosides. Ionograms of individual ginsenoside standards at different gas modifier concentrations ranging from 0 to 0.4% were obtained. Mixtures of isomeric ginsenosides at 2.0–3.0 μM in methanol/water (50:50 v/v + 0.1% sodium acetate) were used in the DMS-MS/MS experiments. For quantitative DMS-MS/MS analysis, calibration plots of the relative peak areas of the DMS-MS/MS ionograms were obtained using mixtures of isomeric ginsenosides. Mixtures of isomeric ginsenosides (Rg1/Re, Rb1/Rg1, and Rb2/Rc) with incremental ratios of 1:9; 3:7; 5:5; 7:3, and 9:1 at a total concentration of 5 μM in methanol/water (50:50 v/v + 0.1% sodium acetate) were analyzed in triplicate [27,28,29,30]. The relative peak areas of the DMS-MS/MS ionograms were plotted against the relative solution concentrations of the mixtures of ginsenosides, i.e., Rg1/Re, Rb1/Rg1, and Rb2/Rc, to obtain the calibration plots.
Results and Discussion
Eight ginsenosides (Rb1, Rb2, Rb3, Rc, Rd., Re, Rf, and Rg1) and a pseudo-ginsenoside (F11), which constitute the major bioactive components in ginseng, were studied in this work. The structural information for these ginsenosides/pseudo-ginsenoside is summarized in Figure 1. All ginsenosides have a common four-ring steroid-like structure but differ in the nature, number, and position of the glycan units. Rb2/Rb3/Rc, Rd./Re, and Rf/Rg1 are three sets of isomeric ginsenosides, and F11 is a pseudo-ginsenoside that is isomeric to Rf/Rg1.

Structures of the ginsenoside (a) Rb1, Rb2, Rb3, Rc, Rd., Re, Rf, and Rg1 and the pseudo-ginsenoside (b) F11
Tandem Mass Spectrometry of the Ginsenosides and Pseudo-ginsenoside
In the presence of a large molar excess of sodium acetate, micro-electrospray ionization (μESI) of the ginsenoside/pseudo-ginsenoside standards in positive mode generated exclusively sodiated molecular ions. The exact mass measurements of these ions were m/z 1131.5922 for Rb1, m/z 1101.5816 for Rb2, Rb3, and Rc, m/z 969.5368 for Rd. and Re, and m/z 823.4807 for Rf, Rg1, and F11. The collision-induced dissociation (CID) of these ginsenoside/pseudo-ginsenoside molecular ions were individually experimentally optimized and their tandem mass spectra were acquired. The CID of the sodium ion adduct of the ginsenoside species yielded a limited number of fragment ions. The predominant dissociation pathways for these sugar-containing compounds were primarily related to the cleavage of glycosidic linkages (Figure S-1 and Table S-1). The loss of disaccharide units from the sodium ion adducts of Rf and F11 resulted in diagnostic fragment ions at m/z 365.1053 and 497.3606, respectively. The loss of one of the sugar substituents from the sodium ion adduct of Rg1 resulted in a diagnostic Z0α/β fragment ion at m/z 643.4175. Cleavage of a glycosidic linkage in the sodium ion adduct of Rb1 resulted in a pair of complimentary fragment ions at m/z 789.4763 and 365.1059. The CID of the other two sets of isomeric ginsenosides, i.e., Rd./Re and Rb2/Rb3/Rc, did not generate any diagnostic fragment ions for their differentiation. A previous study demonstrated that electron-induced dissociation (EID) could provide more structural information and generate some diagnostic fragment ions [5]. However, these diagnostic fragment ions were low in abundance and could not be used for quantitative analysis. To assess the quantitative ratios involving Rd., Re, Rb2, Rb3, and Rc, prior separation of these isomeric ginsenosides is therefore a prerequisite.
Separation of Ginsenoside/Pseudo-ginsenoside Isomers Using DMS-MS/MS
Figure 2a shows three sets of overlaid ionograms for different groups of ginsenoside/pseudo-ginsenoside isomers obtained under the standard DMS conditions using a dispersion field of 127 Td without any gas modifier. Under this moderately high dispersion field, only F11/Rf and Rf/Rg1 pairs could be partially separated, and the other ginsenoside pairs showed only a single peak. The centroids of the main peaks of Rg1, Rf, and F11 were found at around − 1.0 V, whereas the centroids of the Rb2, Rb3, Rc, Rd, and Re peaks were found at around − 2.5 V. All peaks had a full width at half maximum (FWHM) of ~ 1.0 V. As can be seen from the overlaid ionograms, none of the pairs of isomeric ginsenoside ions could be resolved by DMS without the addition of gas modifier. The use of a higher dispersion field (data not shown) did not improve the resolving power.

DMS ionograms of ginsenoside/pseudo-ginsenoside isomers in the presence of (a) 0% 1-propanol and (b) the optimized concentration of 1-propanol as the gas modifier. (c) DMS resolution (resolution = 1.18 × (CoV1− CoV2)/(FWHM1 + FWHM2)) of the ginsenoside/pseudo-ginsenoside isomers using different concentrations of 1-propanol
In a systematic study of a homologous series of n-alcohols (C1-C6) as gas modifiers for DMS analysis of reserpine molecular ions, 1-propanol was found to give the best performance with an accessible vapor pressure, relatively low reduction of the analyte ion signal, and large induced CoV shift (detailed discussion of this experimental result is beyond the scope of this paper, and will be presented in a separate paper). In this study, the possibility of using 1-propanol as the gas modifier to resolve isomeric ginsenoside ions was evaluated. A series of DMS-MS ionograms for each ginsenoside species were obtained using different loadings of 1-propanol (0–0.4%) in the nitrogen carrier gas (Figure S-2). In all cases, the presence of 1-propanol in the carrier gas caused the ionogram peaks to shift towards more positive compensation voltage. Based on the present dispersion and compensation voltages, a positive shift in the compensation voltage implies a reduction in the low field mobility of the ion or an enhancement in the high field ion mobility. Considering the dynamic clustering and declustering model [17], 1-propanol might interact with the ginsenoside ions during the low field portion of the cycle, and thereby reducing their mobility. Under the high field, the mobility of the ginsenoside ions should not be significantly affected, presumably due to the disruption of the interaction between 1-propanol and the ginsenoside ions by the strong electric field. Although the ionograms of all the ginsenoside ions were shifted to more positive CoV, differences in the magnitude of the shift at certain 1-propanol loadings were observed. This differential shift led to the nearly baseline resolution of all the isomeric pairs of ginsenoside ions, except Rb2/Rb3/Rc. Figure 2b shows the three sets of overlaid ionograms for the different groups of isomeric ginsenosides obtained under the same DMS conditions with the addition of 1-propanol as the gas modifier. The optimum separations of Rd/Re and Rg1/F11/Rf were achieved at 1-propanol loadings of 0.08 and 0.13%, respectively. For Rb2/Rb3/Rc set, there was no optimum 1-propanol loading for complete separation of these three isomeric species. From a systematic investigation of the CoV shifts of these ginsenosides, it was discovered that Rb3 could be well separated from Rb2/Rc at 1-propanol loading of 0.13%, whereas Rb2 could be separated from Rb3/Rc at 1-propanol loading of 0.33% (see Figure S-3 for a plot of the CoV maximum versus the 1-propanol loading for Rb2, Rb3, and Rc). The corresponding DMS-SRM ionograms under CID conditions are shown in Figure S-4.
Resolving Power Versus 1-Propanol Loading
Figure 2c shows a plot of the predicted resolving powers of several pairs of isomeric ginsenosides versus the 1-propanol loadings. The predicted resolving powers were computed from the peak position (CoVmax) and the FWHM of the corresponding pair of isomeric ginsenosides ions (Table S-2). Intriguingly, these pairs of isomeric ginsenoside ions were largely overlapped in the absence of the modifier 1-propanol, but were separated at intermediate loadings of 1-propanol. This finding implies that the structural differences among the isomeric ginsenosides could be amplified through interactions with 1-propanol in the gas phase. Among these sets of isomeric ginsenosides, the results obtained for Rb2/Rb3/Rc deserve special attention. Rb2, Rb3, and Rc have high structural similarity, differing only in one of the sugar moieties in the R3 substituent (see Figure 1), i.e., Rb2 has an arabinopyranose (the hydroxyl group at 4-carbon is in axial position), Rb3 has a xylose (the hydroxyl group at 4-carbon is in equatorial position), while Rc has an arabinofuranose. Arabinopyranose in Rb2 differs from xylose in Rb3 only at the C4-carbon where the hydroxyl group is positioned in axial position in arabinopyranose but in equatorial position in xylose. From Figure 2c, it was intriguing to noted that Rb2 and Rb3 could be separated upon addition of small amount of 1-propanol modifier (i.e., > 0.1%). From a thermodynamic point of view, 1-propanol should interact preferentially at the charging site because the ion-dipole interaction should be the strongest among the various available modes of intermolecular interaction. Therefore, the sodium ion binding sites of both of the isomeric ginsenosides Rb2 and Rb3 should be located in the vicinity of the R3 substituents. Interactions between the 1-propanol molecule(s) and the sodiated ginsenosides would be significantly influenced by the surrounding environment of the charge center leading to a differential shift in the optimal compensation voltage for transmission of these two ginsenoside isomeric ions. For the isomer Rc, the anabinofuranose has a five-membered ring structure as opposed to the six-membered ring structures in arabinopyranose and xylose. It was therefore tentative suggested the similar shifts in the optimal CoV for Rb2 and Rc at low to medium 1-propanol loading (0–0.2%) were co-incidental. It was unfortunately that the optimal CoV for Rc was found to shift closer to that of Rb3 at relatively high loading of 1-propanol (0.2–0.4%). Unlike the other two sets of structurally different isomeric ginsenosides (Rg1/F11/Rf and Rd/Re), no optimal 1-propanol loading could be obtained for the separation of these three isomeric ginsenosides ions.
Quantitative DMS-MS/MS Analysis for Ginseng Samples
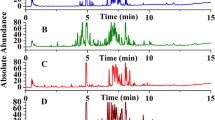
Having established unique experimental conditions based on the 1-propanol loadings for the fast separation of these groups of isomeric ginsenosides, an attempt was made to derive an acquisition protocol for ginseng extract analysis based on DMS-MS analysis using the stepwise modulation of 1-propanol loadings. Figure 3 shows selected-ion ionograms of the ginseng extracts obtained at three different 1-propanol loadings (0.08% for m/z 969.5368; 0.13% for m/z 823.4807; and 0.33% for m/z 1101.5816). After each increment in the 1-propanol concentration, a 2-min delay time was applied to allow the 1-propanol concentration to stabilize. Six ginseng samples, including three ginsengs from Northern America and three ginsengs from Asian countries, were obtained commercially and were extracted and purified using previously established methods [3]. Three ginseng samples were whole ginseng, and three ginseng samples were sliced products. Figure 3 clearly demonstrates that the almost baseline resolution of the three groups of isomeric ginsenosides was retained in the analysis of ginseng sample extracts. The assignments of the ionogram peaks were based on a comparison of the measured CoV values to those of the reference standards. Figure 3b shows unambiguously that the American ginseng samples (USA whole and sliced ginseng and Canadian sliced ginseng) contained an abundant amount of pseudo-ginsenoside F11 with little or no ginsenoside Rf, whereas the Asian ginseng samples (Chinese whole and sliced ginsengs and Korean whole ginseng) contained some Rf but no detectable F11.

DMS ionograms of the ginsenoside/pseudo-ginsenoside isomers of six ginseng extracts obtained using different amounts of 1-propanol as the gas modifier: (a) 0.08% for Rd and Re, (b) 0.13% for Rg1, Rf, and F11, and (c) 0.33% for (Rb3 + Rc) and Rb2
The sensitivity of the DMS-MS/MS-based method for quantification of the ginsenosides used in the present study was also tested. DMS experiments were operated with 0.08% (Re and Rd), 0.13% (Rf, Rg1, F11, Rb2, Rc, Rb3), and 0.33% (Rb2, Rb3, and Rc) 1-propanol as gas modifier, respectively. The standard calibration curves were obtained by different concentrations of ginsenoside mixture over the linear range between 0.01–0.05 μM and 10 μM (0.01, 0.025, 0.05, 0.1, 0.5, 1.0, 5.0, and 10.0 μM) (Figure S-6). The error bars were obtained from the standard deviations of three replicate measurements. The limits of quantification (LOQs) in DMS-SRM for nine ginsenosides/pseudo-ginsenoside Rf, Rg1, F11, Rd, Re, Rb2, Rb3, Rc, and Rb1 were 0.01 μM (8.0 ng/mL), 0.01 μM (8.0 ng/mL), 0.025 μM (20.0 ng/mL), 0.05 μM (47.5 ng/mL), 0.05 μM (47.5 ng/mL), 0.025 μM (27.0 ng/mL), 0.01 μM (10.8 ng/mL), 0.05 μM (54.0 ng/mL), and 0.05 μM (55.0 ng/mL), respectively.
It has been proposed that the relative amounts of several pairs of ginsenosides (i.e., Rg1/Re, Rb1/Rg1 and Rb2/Rc) can be used to differentiate American and Asian ginseng [12]. The use of a single parameter (i.e., either F11 or Rf) to differentiate American and Asian ginseng is simple and convenient, but a more elaborate method based on the benchmark ratios of selected ginsenosides can provide higher confidence of authentication. Although only Rb2/Rb3/Rc are isomers in these pairs of ginsenosides, measurement of the ratios of Rg1/Re and Rb1/Rg1 requires these ginsenosides to be distinguishable from their isomeric counterparts. To quantify Re without interference from Rd., the measurement must be conducted at a 1-propanol loading of 0.08%. However, at this 1-propanol loading, Rg1 would not be completely separated from F11. Since Rg1 and F11 have quite different MS/MS spectra, Rg1 could easily be quantified without interference from F11 using selected reaction monitoring (SRM). To obtain Rb2/Rc ratio, it was necessary to eliminate the contribution of Rb3. In the present study, the ratio of Rb3 to the sum of Rb2 and Rc was determined from the ionogram obtained at 0.13% 1-propanol loading. This ratio was then used to back calibrate the relative area of Rb2 and Rc from the ionogram obtained at 0.33% 1-propanol loading using their calibration plots obtained at both 0.13 and 0.33% 1-propanol loadings (see Figure S-5 and Table S-3).
Table 1 summarizes that relative peak areas of Rg1/Re, Rb1/Rg1, and Rb2/Rc obtained from the SRM chromatograms of the six ginseng extracts obtained at 0.08, 0.13, and 0.33% 1-propanol loadings, respectively. To obtain quantitative ratios, the instrument was calibrated using mixtures of the ginsenoside standards at different molar ratios at the corresponding 1-propanol loadings (i.e., Rg1/Re at 0.08% 1-propanol, Rb1/Rg1 at 0.13% 1-propanol, and Rb2/Rc at 0.33% 1-propanol). Representative SRM ionograms are shown in Figure S-6. Figure 4 shows the calibration plots for (a) Rg1/Re, (b) Rb1/Rg1, and (c) Rb2/Rc. The error bars were obtained from the standard derivations of three replicate measurements. In all cases, good linearity (R2 ≥ 0.995) was obtained between the area ratios and the concentration ratios.

Calibration plots of the relative peak areas of the DMS-MS/MS ionograms and the relative solution concentrations of the three sets of ginsenosides: (a) Rg1/Re, (b) Rb1/Rg1, and (c) Rb2/Rc. The error bars were obtained from the standard deviations of three replicate measurements. The relative peak areas of the three pairs of ginsenosides extracted from the six ginseng extracts were fitted along with the regression lines
Benchmark Cutoffs for Differentiating American and Asian Ginseng
To visualize the clustering of the data obtained from the American and Asian ginseng samples, the experimental peak area ratios for all the ginseng extracts were plotted onto the regression line of the calibration plots. The calculated concentration ratios for these pairs of ginsenosides were extracted and are listed in Table 1. The Rg1/Re and Rb2/Rc ratios of the American ginseng samples were significantly smaller than those of the Asian ginseng samples, whereas the Rb1/Rg1 ratios of the American ginseng samples were much higher than those of the Asian ginseng samples. Qi and co-workers proposed benchmark ratios for American ginseng of Rg1/Re < 1.0, Rb1/Rg1 > 5.0, and Rb2/Rc < 0.4 [12]. Our experimental Rb1/Rg1 and Rb2/Rc ratios show good agreement with these benchmark values. For the Rg1/Re ratio, all the ginseng samples showed experimental values (American ginseng samples ≤ 0.1; Asian ginseng samples ≤ 0.9) that were lower than that of the benchmark value for American ginseng of < 1.0, even though the obtained values from the American ginseng extracts were significantly lower than those obtained from Asian ginseng extracts.
This discrepancy might be related to the difference in the way in which the ginseng samples were prepared and extracted. In the present study, whole ginseng, including the main root and root hairs, was homogenized and extracted. It has previously been reported that the content of the ginsenoside Re in the root hair of Asian ginseng is ten times higher than that of the main root [31]. Therefore, the inclusion of root hairs in the sample might lead to a reduction in the Rg1/Re ratio. In addition, the readings obtained from the sliced products show some differences as compared to those of the whole ginseng samples. It is believed that the short steaming or heating of the dried ginseng required by the slicing of the ginseng might cause some changes in the ginsenoside profiles [32]. Nevertheless, distinctive differences between American and Asian ginseng samples could still be observed in the quantitative ratios of these pairs of ginsenosides.
In order to account for the subtle differences induced by the ginseng processing, new benchmark cutoffs for differentiating American and Asian ginseng were proposed using the data obtained in this study. The cutoff (X) for a particular pair of ginsenosides should be related to the mean (μ) and the standard deviation (σ) of the ratios of that pair of ginsenosides obtained from various American ginseng samples (or from Asian ginseng samples):
where α is an adjustment factor, n is the number of samples, where n1, n2 = 3, and X was calculated by solving α. The new cutoffs are shown as vertical dotted lines in Figure 4. The new benchmarks for American ginseng are Rg1/Re < 0.15, Rb1/Rg1 > 2.15, and Rb2/Rc < 0.26.
There are several advantages for using DMS rather than UHPLC for separation of these isomeric ginsenosides. The total acquisition time used in the developed protocol is 8.5 min (including already 2 × 2-min delay for stabilization of the organic modifier concentration after each stepwise increment). This is comparable to or even faster than the conventional UHPLC method [33, 34]. In addition, no prolong flushing of the system is needed between batches of analysis. No high cost UHPLC column is needed. The solvent used in DMS experiment is quite minimal (~ 30 μL per experiment) as compared to UHPLC. The home-built DMS system costs roughly one-tenth of the cost of a commercially available UPLC system.
Conclusions
A DMS-MS/MS-based method was established for the separation of ginsenoside/pseudo-ginsenoside isomers and for the differentiation of Asian and American ginseng. Our results indicate that the amount of the modifier is crucial to achieving good separation of isomeric ginsenosides. This is the first work using DMS with stepwise modulation of gas modifier concentration to assist the separation of isomers and increase the resolving power of DMS. The quantitative aspect of the DMS-MS/MS method was assessed using the ratios of the signals of three pairs of ginsenosides, namely, Rg1/Re, Rb1/Rg1, and Rb2/Rc. Good linearity between the signal ratios and the concentration ratios was obtained. Six commercially available ginseng samples, including three American ginseng and three Asian ginseng samples, were successfully differentiated using this DMS-MS/MS method. Although there were some discrepancies between the obtained ginsenoside ratios and literature benchmark ratios, the ratios obtained from the American and Asian ginseng samples were clustered in different ranges. Based on our results, new benchmark ratios of Rg1/Re < 0.15, Rb1/Rg1 > 2.15, and Rb2/Rc < 0.26 were proposed for American ginseng.
References
Leung, K.W., Wong, A.S.T.: Circumvention of multi-drug resistance of cancer cells by Chinese herbal medicines. Chi. Med. 5, 20–26 (2010)
Baek, S.-H., Bae, O.-N., Park, J.H.: Recent methodology in ginseng analysis. J. Ginseng Res. 3, 119–134 (2012)
Chan, T.W.D., But, P.P.H., Cheng, S.W., Kwok, I.M.Y., Lau, F.W., Xu, H.X.: Differentiation and authentication of Panax ginseng, Panax quinquefolius, and ginseng products by using HPLC/MS. Anal. Chem. 72, 1281–1287 (2000)
Deng, G.F., Wang, D.L., Meng, M.X., Hu, F., Yao, T.W.: Simultaneous determination of notoginsenoside R1, ginsenoside Rg1, Re, Rb1 and icariin in rat plasma by ultra-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B. 877, 2113–2122 (2009)
Wong, Y.L.E., Chen, X.F., Li, W., Wang, Z., Hung, Y.L.H., Wu, R., Chan, T.W.D.: Differentiation of isomeric ginsenosides by using electron-induced dissociation mass spectrometry. Anal. Chem. 88, 5590–5594 (2016)
Chen, X.F., Wang, Z., Wong, Y.L.E., Wu, R., Zhang, F., Chan, T.W.D.: Electron-ion reaction-based dissociation: a powerful ion activation method for the elucidation of natural product structures. Mass Spec. Rev. 37, 793–810 (2018)
Song, F.R., Liu, Z.Q., Liu, S.Y., Cai, Z.W.: Differentiation and identification of ginsenoside isomers by electrospray ionization tandem mass spectrometry. Anal. Chim. Acta. 531, 69–77 (2005)
Vanhaelen-Fastré, R.J., Faes, M.L., Vanhaelen, M.H.: High-performance thin-layer chromatographic determination of six major ginsenosides in Panax ginseng. J. Chromatogr. A. 868, 269–276 (2000)
Cui, J.F., Björkhem, I., Eneroth, P.: Gas chromatographic-mass spectrometric determination of 20(S)-protopanaxadiol and 20(S)-protopanaxatriol for study on human urinary excretion of ginsenosides after ingestion of ginseng preparations. J. Chromatogr. B. 689, 349–355 (1997)
Ren, G.X., Chen, F.: Simultaneous quantification of ginsenosides in American ginseng (Panax quinquefolium) root powder by visible/near-infrared reflectance spectroscopy. J. Agric. Food Chem. 47, 2771–2775 (1999)
Akao, T., Kanaoka, M., Kobashi, K.: Appearance of compound K, a major metabolite of ginsenoside Rb1 by intestinal bacteria, in rat plasma after oral administration: measurement of compound K by enzyme immunoassay. Biol. Pharm. Bull. 21, 245–249 (1998)
Qi, L.W., Wang, C.Z., Yuan, C.S.: Ginsenosides from American ginseng: chemical and pharmacological diversity. Phytochemistry. 72, 689–699 (2011)
Dodds, J.N., May, J.C., McLean, J.A.: Correlating resolving power, resolution and collision cross section: unifying cross platform assessment of separation efficiency in ion mobility spectrometry. Anal. Chem. 89, 12176–12184 (2017)
Li, H.L., Giles, K., Bendiak, B., Kaplan, K., Siems, W.F., Hill, H.H.: Resolving structural isomers of monosaccharide methyl glycosides using drift tube and traveling wave ion mobility mass spectrometry. J. Anal. Chem. 84, 3231–3239 (2012)
Regueiro, J., Giri, A., Wenzl, T.: Optimization of a differential ion mobility spectrometry-tandem mass spectrometry method for high-throughput analysis of nicotine and related compounds: application to electronic cigarette refill liquids. Anal. Chem. 88, 6500–6508 (2016)
Purves, R.W., Ozog, A.R., Ambrose, S.J., Prasad, S., Belford, M., Dunyach, J.J.: Using gas modifiers to significantly improve sensitivity and selectivity in a cylindrical FAIMS device. J. Am. Soc. Mass Spectrom. 25, 1274–1284 (2014)
Levin, D.S., Vouros, P., Miller, R.A., Nazarov, E.G., Morris, J.C.: Characterization of gas-phase molecular interactions on differential mobility ion behavior utilizing an electrospray ionization-differential mobility-mass spectrometer system. Anal. Chem. 78, 96–106 (2006)
Kafle, A., Coy, S.L., Wong, B.M., Fornace, A.J.J., Glick, J.J., Vouros, P.: Understanding gas phase modifier interactions in rapid analysis by differential mobility-tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 1098–1113 (2014)
Liu, C., Ríos, G.A.G., Schneider, B.B., Blanc, J.C.Y.L., Garcés, N.R., Arnold, D.W., Covey, T.R., Pawliszyn, J.: Fast quantitation of opioid isomers in human plasma by differential mobility spectrometry/mass spectrometry via SPME/open-port probe sampling interface. Anal. Chim. Acta. 991, 89–94 (2017)
Campbell, J.L., Yang, A.M.C., Melo, L.R., Hopkins, W.S.: Studying gas-phase interconversion of tautomers using differential mobility spectrometry. J. Am. Soc. Mass Spectrom. 27, 1277–1284 (2016)
McCooeye, M., Ding, L.Y.G., Gardner, J., Fraser, C.A., Lam, J., Sturgeon, R.E., Mester, Z.: Separation and quantitation of the stereoisomers of ephedra alkaloids in natural health products using flow injection-electrospray ionization-high field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal. Chem. 75, 2538–2542 (2003)
Bylda, C., Thiele, R., Kobold, U., Bujotzek, A., Volmer, D.A.: Rapid quantification of digitoxin and its metabolites using differential ion mobility spectrometry-tandem mass spectrometry. Anal. Chem. 87, 2121–2128 (2015)
Chen, P.S., Chen, S.H., Chen, J.H., Haung, W.Y., Liu, H.T., Kong, P.H., Yang, O.H.Y.: Modifier-assisted differential mobility-tandem mass spectrometry method for detection and quantification of amphetamine-type stimulants in urine. Anal. Chim. Acta. 946, 1–8 (2016)
Baba, T., Campbell, J.L., Blanc, J.C.Y.L., Baker, P.R.S., Ikeda, K.: Quantitative structural multi-class lipidomics using differential mobility-electron impact excitation of ions from organics (EIEIO) mass spectrometry. J. Lipid Res. 59, 910–919 (2018)
Wu, R., Wu, W.J., Wang, Z., Wong, Y.L.E., Hung, Y.L.W., Wong, H.T., Chen, X.F., Chan, T.W.D.: Performance enhancements in differential ion mobility spectrometry-mass spectrometry (DMS-MS) by using a modified CaptiveSpray source. J. Am. Soc. Mass Spectrom. 29, 2199–2207 (2018)
Domon, B., Costello, C.E.: A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 (1988)
Zhang, J.D., Kabir, K.M.M., Lee, H.E., Donald, W.A.: Metal-ion free chiral analysis of amino acids as small as proline using high-definition differential ion mobility mass spectrometry. Int. J. Mas. Spectrom. Fastre. 428, 1–7 (2018)
Mie, A., Ray, A., Axelsson, B.O., Karlsson, M.J., Reimann, C.T.: Terbutaline enantiomer separation and quantification by complexation and field asymmetric ion mobility spectrometry-tandem mass spectrometry. Anal. Chem. 80, 4133–4140 (2008)
Mie, A., Karlsson, M.J., Axelsson, B.O., Ray, A., Reimann, C.T.: Enantiomer separation of amino acids by complexation with chiral reference compounds and high-field asymmetric waveform ion mobility spectrometry: preliminary results and possible limitations. Anal. Chem. 79, 2850–2858 (2007)
Zhang, J.D., Kabir, K.M.M., Donald, W.A.: Chiral recognition of amino acid enantiomers using high-definition differential ion mobility mass spectrometry. Anal. Chim. Acta. 1036, 172–178 (2018)
Kang, O.J., Kim, J.S.: Comparison of ginsenoside contents in different parts of korean ginseng (Panax ginseng C.A. Meyer). Prev. Nutr. Food Sci. 21, 389–392 (2016)
Wang, C.Z., Aung, H.H., Ni, M., Wu, J.A., Tong, R., Wicks, S., He, T.C., Yuan, C.S.: Red American ginseng: ginsenoside constituents and antiproliferative activities of heat-processed Panax quinquefolius roots. Planta Med. 73, 669–674 (2007)
Liang, Z.T., Chen, Y.J., Xu, L., Qin, M.J., Yi, T., Chen, H.B., Zhao, Z.J.: Localization of ginsenosides in the rhizome and root of Panax ginseng by laser microdissection and liquid chromatography-quadrupole/time of flight-mass spectrometry. Pharmaceut. Biomed. 105, 121–133 (2015)
Song, Y.L., Zhang, N., Jiang, Y., Li, J., Zhao, Y.F., Shi, S.P., Tu, P.F.: Simultaneous determination of aconite alkaloids and ginsenosides using online solid phase extraction hyphenated with polarity switching ultra-high performance liquid chromatography coupled with tandem mass spectrometry. RSC Adv. 5, 6419–6428 (2015)
Acknowledgements
This work was supported by The National Key Research and Development Program of China (2018YFC1603500), Research Grant Council of the Hong Kong Special Administrative Region (Research Grant Direct Allocation, Ref. 3132667 and 4053152), Key R&D Program of Shandong Province (2019GSF111009, 2019GSF111001), and Natural Science Foundation of Shandong Province (ZR2017MB011).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Electronic Supplementary Material
ESM 1
(DOCX 1234 kb)
Rights and permissions
About this article

Cite this article
Wu, R., Chen, X., Wu, WJ. et al. Rapid Differentiation of Asian and American Ginseng by Differential Ion Mobility Spectrometry-Tandem Mass Spectrometry Using Stepwise Modulation of Gas Modifier Concentration. J. Am. Soc. Mass Spectrom. 30, 2212–2221 (2019). https://doi.org/10.1007/s13361-019-02317-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02317-0